TULBURĂRILE HEMOSTAZEI
Hemostaza este rapunsul organismului la agresiuni vasculare. Ea implica participarea vasului, a placutelor si a sistemului coagulare-fibrinoliza în echilibru fin reglat.
Factorul declansator al activarii hemostazei este pierderea functiei de bariera a endoteliului vascular (separa fluxul sanguin de spatiul subendotelial). În mod arbitrar raspunsul plachetar poate fi împartit în trei etape: adeziune, activare si agregare. Rezultatul este formarea cheagului plachetar.
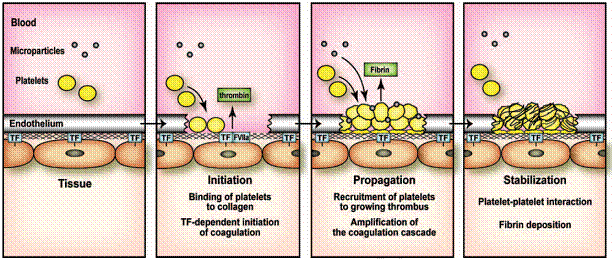
Figura 1
Factorii tisulari (TF) de la nivelul leziunii vasculare activeaza simultan si sistemul coagularii, ceea ce duce la formarea fibrinei la nivelul leziunii.
Tot acest proces este fin controlat de modulatorii placutelor, reactivitatii vasculare (NO, prostaciclina) si ai coagularii (antitrombina III, sistemul proteinei C, inhibitori ai factorilor tisulari), pentru a limita tromboza. Astfel, procoagulantii sunt diluati si îndepartati de la nivelul leziunii de fluxul sanguin, factorii coagularii activati sunt degradati la nivelul sistemului reticuloendotelial, iar fibrinoliza îndeparteaza cheagul de fibrina.
Tulburarile hemostazei pot fi grupate în sindroame hemoragice si sindroame trombotice.
SINDROAMELE HEMORAGICE
Principalele caracteristici ale sindroamelor hemoragice
|
|
Localizarea hemoragiei |
|
|
|||
|
Tip de tulburare |
General |
Tegument |
Mucoase |
Alte |
Debut |
Exemple |
|
Tulburari p 11511r171l lachetare si vasculare |
Superficial |
Petesii, echimoze |
Frecvent: oral, nazal, gastrointestinal, genitourinar |
Rare |
Spontane sau imediat dupa traume |
Trombocitopenia, trombocitopatii, fragilitate vasculara, CID, boli vasculare |
|
Deficit de factori ai coagularii |
Profound |
Hematoame |
Rare |
Frecvent : articulatii, muschi, retroperitoneal |
Cu întârziere dupa traume |
Deficite ereditare ale factorilor coagularii, inhibitori dobânditi, anticoagulanti, CID, boli hepatice |
ANOMALIILE PLĂCUŢELOR
Sindroamele hemoragice determinate de placute pot fi cauzate de scaderea numarului de placute (trombocitopenie) sau de tulburari functionale plachetare (trombocitopatii). Ambele tipuri pot avea cauze ereditare sau dobândite.
TROMBOCITOPENIA
Trombocitopenia poate fi determinata de:
Cresterea distructiei sau utilizarii placutelor
Distributie anormala a placutelor
Reducerea producerii placutelor.
Presupunând ca functia este normala, clinic exista o corelatie între reducerea numarului de placute si sindromul hemoragic :
CREsTEREA DISTRUCŢIEI SAU UTILIZĂRII PLĂCUŢELOR
Distructie imuna
DISTRIBUŢIE ANORMALĂ A PLĂCUŢELOR
TAI cronica este o afectiune autoimuna. Nu se cunoaste factorul ce declanseaza reactia autoimuna, dar au fost identificati autoanticorpi antiplachetari. Placutele acoperite de autoanticorpi sunt distruse prin fagocitoza sau de complement. Acuzele sunt de cel putin 6 luni.
Purpura trombocitopenica autoimuna acuta
TAI acuta este tranzitorie, autolimitanta, cel mai frecvent postinfectioasa (viroze), si apare aproape exclusiv la copilul mic (2-5ani). Ea se datoreaza unui raspuns autoimun comparabil cu cel din PTIC.
Purpura trombocitopenica autoimuna acuta asociata unor alte boli primare
Cel mai frecvent a fost constatata asocierea cu bolile de colagen (LED) si bolile limfoproliferative leucemia limfocitica cronica, limfom non-Hodgkin).
Trombocitopenia aloimuna
Cel mai frecvent apare post-transfuzional la femei cu transfuzii în antecedente sau la multipare la prima transfuzie. Placutele pacientelor nu au aloantigenele, dar aloanticorpii reactioneaza cu glicoproteine de membrana. Exista trei ipoteze de mecanism:
Purpura trombocitopenica imuna indusa de medicamente
Datorita greutatii moleculare mici, medicamentele ce se fixeaza pe placute actionaza ca haptene ( chinidina, chinina, sulfonamidele, sarurile de aur, antagonisti ai GPIIb-IIIa plachetare) si pot determina distructia placutelor prin producerea de autoanticorpi fata de neoantigenele astfel formate.
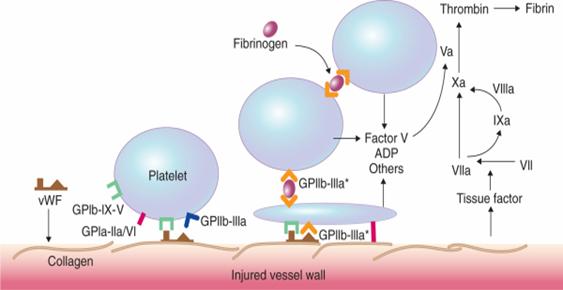
Figura 2
Trombocitopenia indusa de heparina (TIH)
TIH se datoreaza sintezei de autoanticorpi împotriva neoantigenelor formate prin legarea heparinei de proteina cationica alfa2-globulina.
Distructia nonimuna a placutelor
Purpura trombotica trombocitopenica Moskovitz (PTT)
Este o boala foarte rara, care apare la tineri. Debuteaza acut. Mecanismul este o agregare diseminata reversibila a placutelor si tromboze în mica circulatie, datorita unui defect ereditar, congenital sau dobândit de clivaza (ADAMTS-13) a multimerilor de vWF. Rezultatul este trombocitopenie prin consum, ischemie tisulara prin tromboze (insuficienta renala, hipoxie cerebrala) si hemoliza. Mortalitatea este foarte crescuta.
Sindromul hemolitic uremic Gasser
Clinic este similar PTT, afecteaza în special copiii si se manifesta predominant în insuficienta renala. Mecanismul este diferit. Nu au deficit de ADAMTS-13, ci apar leziuni endoteliale, la copii determinate de toxinele din infectiile gastrointestinale cu Escherichia coli, la adult dupa anumite medicamente, postpartum, dupa tratament imunosupresor, transplant de maduva sau de organe. Formele familiale sunt rare.
Coagularea intravasculara diseminata
Numeroase afectiuni (infectii, arsuri, traumatisme, neoplasme) se însotesc de eliberare masiva de procoagulanti în circulatia sistemica, astfel ca mecanismele de control sunt depasite si formarea de trombina duce la coagularea intravasculara diseminata. Rezultatul final este aparitia multiplelor disfunctii de organ. În acest proces trombocitopenia apare prin consum crescut.
Trombocitopenia dupa transfuziile masive se datoreaza pierderii de placute prin hemoragie si înlocuirea lor cu placute nefunctionale din produsul transfuzat (prin pastrare la temperaturi scazute functia placutelor se pierde).
Distributia anormala a placutelor
În splenomegalie creste depozitarea placutelor la nivel splenic, si în consecinta scade nivelul lor la nivelul circulatiei sistemice.
Reducerea producerii de placute
Principalele mecanisme de reducere aproducerii de placute sunt:
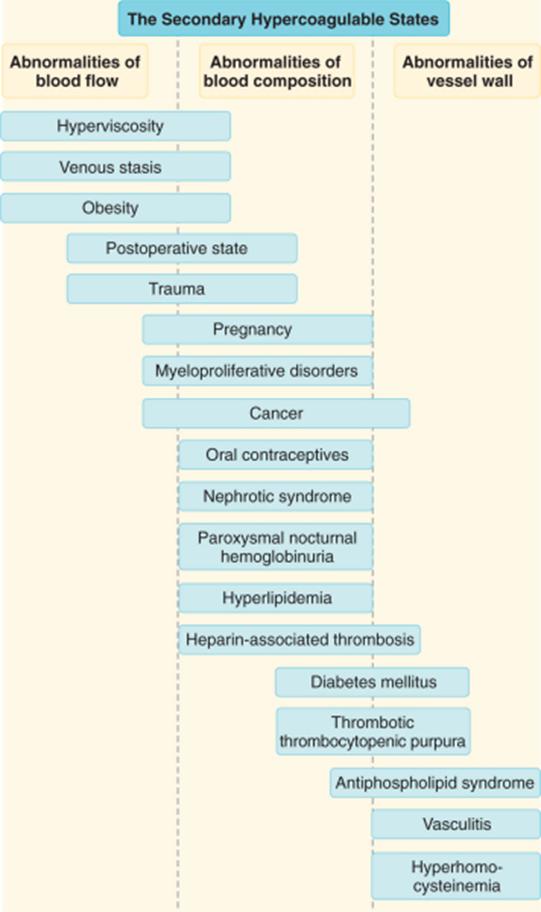
Cele mai frecvente manifestari la pacientii cu neoplasme sunt tromboza venoasa profunda, embolia pulmonara, sindromul Trousseau (tromboflebita superficiala migratorie a membrelor superioare sau inferioare).
Sindromul antifosfolipidic
Se caracterizeaza prin tromboze venoase si arteriale, avort spontan recurent, trombocitopenie, si diferite manifestari neuropsihiatrice. Mecanismul implica formarea unui grup heterogen de autoanticorpi care se leaga de complexe fosfolipid-proteina amniotice.
|
