Cromatografia de lichide
Termenul de cromatografie de lichide este utilizat pentru a denumi o gama extinsa de sisteme cromatografice care au în comun faptul ca utilizeaza o faza mobila lichida.
Comparativ cu cromatografia de gaze, cea de lichide are avantajul ca permite si analizarea probelor greu volatile sau instabile termic, iar faptul ca si faza mobila participa la separare ofera posibilitati suplimentare pentru realizarea unor separari eficiente.
Cromatografia de lichide clasica a fost caracterizata de utilizarea unor coloane cu diametre mari, umplute cu particule de faza stationara de o granulatie fina si prin care faza mobila trecea sub influenta fortei gravitationale. Aceste sisteme, care se mai folosesc azi în separarile cromatografice lichid-solid, au permis realizarea separarii unor amestecuri complexe de substante în conditii foarte bune, însa viteza de separare este mica iar analiza suplimentara a compusilor separati prin metode spectroscopice este mai dificila. Aparitia cromatografiei de lichide de înalta performanta (HPLC) a permis eliminarea acestor deficiente si chiar impunerea cromatografiei de lichide ca o metoda mai performanta de analiza decât cromatografia de gaze. Principalele avantaje ale cromatografiei de lichide de înalta performanta sunt:
Capacitate de separare ridicata
Viteza ridicata a separarii
Posibilitatea monitorizarii continue a efluentului coloanei
Realizarea unor analize repetate si reproductibile utilizând aceeasi coloana
Automatizarea procedurii analitice si a prelucrarii datelor.
Primul cromatograf de lichide modern a fost construit de Csaba Horváth la Universitatea din Yale în 1964 si a tehnica fost denumita cromatografie de lichide de înalta presiune (HPLC). În continuare, termenul care s-a impus a fost cel de cromatografie de lichide de înalta performanta. Prima separare realizata de grupul lui Horváth a fost cea a unor componente ale acizilor nucleici. Dezvoltarea extraordinara a acestei tehnici în ultimii zeci de ani se datoreste în mare masura faptului ca a permis analiza unor compusi care puteau fi separati foarte greu pe alte cai, de exemplu a biomoleculelor.
Principalele tipuri de cromatografie de lichide sunt:
cromatografia de adsorbtie lichid-solid (LSC)
cromatografia de repartitie lichid-lichid (LLC)
cromatografia ionica (IC)
cromatografia prin excluziune (SEC)
4.1. Factorii care influenteaza separarea în cromatografia de lichide
Pentru a putea utiliza cromatografia de lichide ca metoda de analiza în conditii satisfacatoare, nu este necesara cunoasterea în amanunt a teorei cromatografiei de lichide, dar este indispensabila stapânirea notiunilor de baza, mai ales pentru a se putea face selectia fazelor cromatografice într-o maniera corecta.
În ciuda complexitatii aparente a instrumentului, partea esen&# 444i86e 355;iala a analizei cromatografice este procesul de separare care are loc în coloana, restul aparaturii având doar rolul auxiliar de a asigura depozitarea fazei mobile, asigurarea compozitiei sale stabilite si a presiunii de intrare în coloana, introducerea probei, detectarea componentelor separate si procesarea datelor. Ca urmare a modului în care este conceput designul aparatelor moderne, aceasta esenta se pierde însa de multe ori în spatele complexitatii electronice sau ingineresti a întregului sistem. Rezultatul este ca rolul important al functiunii coloanei este înteles doar superficial si tehnica cromatografica nu este utilizata într-o maniera adecvata.
În timpul separarii cromatografice în coloana au loc doua procese care se produc simultan, continuu si aparent independent una de alta. Prima este deplasarea fiecarui solut printr-o succesiune de echilibre de distributie între faza mobila si cea stationara, în conformitate cu coeficientul sau de distributie. Al doilea proces este specific coloanei, care trebuie sa limiteze dispersia naturala a solutilor astfel încât la iesirea din coloana fiecare solut sa se regaseasca sub forma unei benzi de elutie cât mai înguste si distincte de celelalte. Primul proces, cel al retentiei componentelor, este de natura termodinamica si poate fi controlat prin alegerea corespunzatoare a naturii fazelor cromatografice. Cel de-al doilea proces este, în esenta, de natura cinetica si depinde de proprietatile fizice ale componentelor sistemului de separare cromatografica: faza stationara si faza mobila. Pentru a putea realiza o separare optima, pentru fiecare proba va trebui gasita perechea de faze cea mai potrivita si selectata coloana corespunzatoare.
În cromatografia de lichide, ecuatia esentiala care permite întelegerea procesului de retentie si identificarea parametrilor cu ajutorul carora va fi posibila optimizarea analizei este ecuatia volumului de retentie.
![]() (60)
(60)
unde VM este volumul mort al coloanei, K este coeficientul de distributie al solutului, iar VS este volumul fazei stationare. Aceasta relatie este dedusa pe baza teorei talerelor a lui Martin si Synge, perfectionata apoi de alti autori. În cromatografia de lichide, spre deosebire de cromatografia de gaze, volumul fazei stationare din coloana nu mai poate fi definit asa de net. În majoritatea cazurilor, în separare nu intervine întregul acest volum ci numai volumul fazei stationare disponibil solutului.
Separarea a doi compusi va fi posibila daca volumele lor de retentie vor fi diferite, adica în cazul în care fie coeficientii de distributie ai celor doi soluti vor fi diferiti, fie volumul de faza stationara disponibil fiecarui component al amestecului analizat va fi diferit. Asadar pentru a realiza separarea dorita trebuie studiati factorii care influenteaza acesti doi parametri, coeficientul de distributie si volumul fazei stationare disponibile.
Coeficientul de distributie este raportul dintre concentratia solutului în faza stationara si concentratia în faza mobila, fiind o masura a interactiunilor moleculare dintre solut si cele doua faze.
Aceste interactiuni sunt consecintele fortelor ce se manifesta între moleculele celor doua faze cromatografice si cele ale solutului Aceste forte pot fi de patru tipuri: forte chimice, forte ionice, forte polare si forte de dispersie. Exista si alte forte, cele bazate pe legaturi de hidrogen, însa pentru simplificare acestea sunt considerate ca fiind o categorie de forte polare foarte puternice. Prin alegerea corespunzatoare a fazelor, toate aceste interactiuni pot fi astfel dirijate încât sa se realizeze retentia dorita.
Fortele chimice pot fi considerate practic ireversibile, cel putin în cromatografie si drept consecinta coeficientul de distributie al solutului legat prin asemenea forte de faza stationara este apropiat de infinit. Un exemplu de cromatografie de lichide bazat pe asemenea interactiuni este cea de afinitate, în care una dintre moleculele probei analizate interactioneaza chimic cu gruparea de afinitate a fazei stationare si substanta respectiva este extrasa selectiv dintre compusii prezenti în sistem. Acest tip de cromatografie reprezinta asadar mai mult o extractie decât o separare cromatografica, iar în celelte tipuri de cromatografie fortele chimice nu au un rol semnificativ.
Fortele ionice se bazeaza pe sarcinile electrice care rezulta în urma ionizarii moleculelor în solutie. Aceste interactiuni sunt exploatate de cromatografia ionica. Daca solutul se prezinta sub forma de anioni, ca de exemplu în cazul acizilor organici, faza stationara va trebui sa contina cationi cu sarcina pozitiva drept contraioni pentru a interactiona cu anionii respectivi si a încetini deplasarea lor prin coloana. În mod corespunzator, pentru separarea compusilor cu caracter cationic prezenti în proba analizata, faza stationara va trebui sa contina contraioni de tip anionic. Fazele stationare ionice sunt constituite în mod obisnuit din polimeri reticulati care au fost modificati chimic pentru a li se grefa gruparile schimbatoare de ioni dorite. O alta alternativa este utilizarea unor faze chimic legate cu grupari schimbatoare de ioni, în acest caz gruparile respective fiind legate chimic de un suport de silicagel, asemenanator ca în cazul altor faze stationare chimic legate. A treia posibilitate este de a adsorbi compusul schimbator de ioni pe suprafata fazei stationare, acest tip de cromatografie fiind denumit cromatografie prin perechi de ioni. Compusul respectiv este introdus în faza mobila în concentratie redusa (în jur de 1%), de unde se adsoarbe pe faza stationara chimic legata.
Fortele polare au la baza de asemenea sarcinile electrice ale moleculelor, însa în acest caz este vorba de dipoli permanenti sau indusi. Trebuie mentionat ca o molecula polara nu poseda sarcini nete, exceptând situatia în care molecula respectiva este în acelasi timp si ionizata. Exemple de substante cu dipoli permanenti sunt alcoolii, esterii, aldehidele, etc. Molecule polarizabile sunt, printre altele, hidrocarburile aromatice ca benzenul, toluenul sau xilenii. Exista compusi cu dipoli permanenti care în acelasi timp sunt si polarizabili, ca de exemplu 2-fenil-etanolul. Pentru a separa compusii polari sau polarizabili, va trebui utilizata o faza stationara polara în combinatie cu o faza mobila relativ nepolara. Realizarea de interactiuni moleculare diferite în cele doua faze reprezinta o modalitate generala pentru a obtine selectivitate în separarile cromatografice. Pentru a realiza retentia si selectivitatea necesara, trebuie ca un anumit tip de interactiuni specifice sa fie dominante în faza stationara, iar interactiunile din faza mobila sa fie diferite fata de cele din faza stationara pentru a mentine selectivitatea fazei stationare. De exemplu pentru a separa niste hidrocarburi aromatice, care sunt compusi polarizabili, se va folosi o faza stationara puternic polara, cum este silicagelul si a faza mobila nepolara, cum este hexanul.
Fortele de dispersie sunt mult mai dificile de caracterizat. Ele sunt în esenta electrice prin natura lor, însa rezulta mai mult din fluctuatiile de sarcina decât din sarcinile electrice propriu-zise ale moleculelor. Exemple de interactiuni de dispersie sunt fortele care se manifesta între moleculele hidrocarburilor saturate. Acesti compusi nu au caracter ionic, nu sunt polarizabile si nu au momente dipol, totusi cele superioare nu sunt gaze ci lichide sau solide întrucât moleculele lor sunt tinute laolalta de fortele de dispersie. Pentru ca fenomenul de retentie sa fie bazat pe interactiuni de dispersie trebuie ca faza stationara sa nu contina grupari ionice sau polare ci numai grupari hidrofobe, asa cum este cazul fazelor stationare inverse din cromatografia HPLC. În plus, faza mobila trebuie sa fie puternic polara pentru ca intercatiunile de dispersie în aceasta faza sa fie minime. Asemenea faze sunt cele de tipul amestecurilor metanol-apa sau acetonitril-apa.
În sistemele reale, doar în putine cazuri este întâlnita situatia în care distributia este determinata doar de un singur tip de interactiuni. Daca totusi se întâmpla acest lucru, poate fi vorba numai despre interactiuni de dispersie. Interactiunile polare sunt însotite întotdeauna de interactiuni ionice si de dispersie, iar cele ionice de interactiuni polare si de dispersie.
Asa cum rezulta din ecuatia volumului de retentie, nu numai taria interactiunii dintre solut si faza stationara va fi cea care determina retentia solutului ci si volumul fazei stationare prezente în sistem si accesibile solutului. Volumul fazei stationare cu care componentele amestecului de analizat pot interactiona depinde de starea fizica a fazei stationare si a suportului. Daca faza stationara este un solid poros si dimensiunile porilor suportului sunt comparabile cu diametrele moleculare ale componentelor de separat, atunci faza stationara va fi selectiva pentru marimea moleculelor acestora. Moleculele anumitor soluti vor putea patrunde în interiorul porilor si vor interactiona cu mai multa faza stationara decât alte molecule mai mari care vor fi partial excluse de la aceste interactiuni. Asadar, separarea va fi controlata de fenomenul de excluziune bazat pe marimea moleculelor si de aceea se numeste cromatografie de excluziune. Daca faza stationara va fi chirala, cantitatea de faza stationara disponibila pentru a interactiona cu solutul va fi doar aceea care din punct de vedere steric se potriveste cu orientarea spatiala a moleculei de solut. Acest tip de cromatografie se numeste cromatografie de lichide chirala. Trebuie mentionat ca în functie de natura fazei stationare ambele aceste tipuri, cromatografia de excluziune sau cromatografia chirala, pot sa se încadreze fie în categoria cromatografiei lichid-lichid fie în a celei de tip lichid-solid.
4.1.3. Eficienta de separare a coloanei cromatografice
În timpul deplasarii prin coloana cromatografica nu este suficient ca benzile de elutie ale celor doi soluti sa migreze diferentiat, ci este necesar ca ele sa fie destul de înguste pentru ca sa se separe cât mai complet una de alta. Parametrul de eficienta care ne arata posibilitatea separarii a doi compusi pe baza selectivitatii termodinamice este coeficientul de separare α, definit pe baza raportului factorilor de capacitate a celor doi soluti.
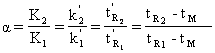 (61)
(61)
Se observa ca coeficientul de separare coreleaza interactiunile din faza stationara si cele din faza mobila.
Asadar în cromatografia de lichide, coeficientul de separare poate fi optimizat prin doua modalitati:
alegerea corespunzatoare a fazei stationare;
alegerea corespunzatoare a fazei mobile.
Toti factorii care modifica interactiunile moleculare sau intensitatea acestora, atât în faza mobila cât si în sau faza stationara vor determina în acelasi timp si modificarea selectivitatii.
În cromatografia de lichide, faza stationara (indiferent ca este polara sau nepolara) va adsorbi întotdeauna în mod preferential o anumita componenta a eluentului, iar grosimea stratului de adsorbtie (df) depinde de natura fazei stationare si a solventului adsorbit (Figura 4.1). Cu cât ne îndepartam de suprafata, cu atât scade taria legaturii acestei adsorbtii si este discutabil unde trebuie plasata delimitarea dintre faza mobila adsorbita si cea neadsorbita. Daca se modifica compozitia fazei mobile acest lucru determina automat si schimbarea compozitiei stratului de faza mobila adsorbita la suprafata fazei stationare, iar drept consecinta modificarea interctiunilor dintre solut si cele doua faze.
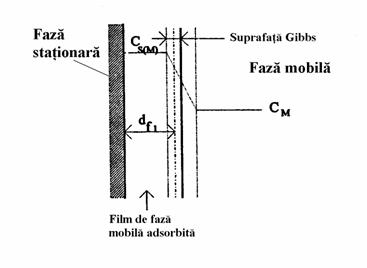
Figura 4.1. Formarea stratului de faza mobila adsorbita pe suprafata fazei stationare
Valoarea calculata a coeficientului de separare depinde de timpul mort al coloanei respective în conditiile de lucru. Determinarea acestui timp mort nu este însa asa de simpla ca în cromatografia de gaze, deoarece într-o coloana cromatografica de gaze volumul mort poate fi net delimitat, în timp ce într-una de lichide aceasta delimitare depinde de grosimea filmului de faza mobila adsorbita la suprafata fazei stationare.
O alta problema în cromatografia de lichide este ca majoritatea adsorbentilor (fazelor stationare) utilizati fiind porosi, într-o parte a porilor faza mobila va stagna si aici compozitia fazei mobile poate fi diferita de cea din zonele unde ea se gaseste în miscare. Pentru a avea valori reproductibile ale timpului mort (tM=L/v), acesta ar trebui determinat folosind un compus care nu interactioneaza cu faza mobila adsorbita. În realitate un asemenea compus nu exista, de aceea întotdeauna trebuie precizat cu ce substanta s-a facut determinarea timpului sau volumului mort. Aceasta relativitate în determinarea timpului mort are influenta si asupra valorilor parametrilor de retentie, mai ales a factorului de capacitate.
Cromatografia de lichide este capabila de rezolvarea unor probleme de separare foarte complexe în buna parte datorita faptului ca prin modificarea sistematica a compozitiei eluentului se pot obtine valori ale coeficientului de separare într-un interval destul de larg.
Realizarea unui coeficient de separare diferit de 1 este o conditie necesara, nu însa si suficienta pentru o separare corespunzatoare, deoarece la aceeasi valoare a acestui coeficient putem avea o separare foarte buna, dar si total nesatisfacatoare a componetelor respective (Figura 4.2).
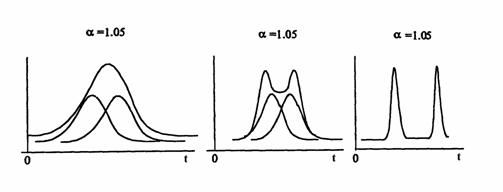
Figura 4.2. Influenta abaterii standard asupra separarii cromatografice
Pentru separarea sa fie corespunzatoare, trebuie ca selectivitatea termodinamica sa fie asociata cu o eficienta cinetica corespunzatoare, adica cu o viteza mare de transfer a moleculelor de solut din faza mobila în faza stationara si invers. Masura acestei eficiente cinetice este latimea picului, respectiv abatarea standard σ. Aceasta abatere standard se poate masura pe baza curbei Gauss care caracterizeaza picul respectiv. Dispersia picului, respectiv cresterea abaterii standard depinde însa nu numai de eficienta cinetica a coloanei ci si de distanta parcursa de solut în coloana, adica de lungimea coloanei. Pentru a se putea face o comparatie între diferite coloane, trebuie utilizata o valoare relativa a dispersiei solutului (si implicit a largirii picului), ceea ce se face cu ajutorul unui parametru de numit numar de talere teoretice N. Valoarea acestuia se calculeaza cu relatia:
![]() (62)
(62)
unde σt si σL reprezinta abaterea standard exprimata în unitati de timp, respectiv în unitati de lungime. Numarul de talere teoretice depinde însa si el de lungimea coloanei, de aceea pentru eliminarea acestor diferente se utilizeaza lungimea de coloana echivalenta unui taler teoretic. Acesta poarta numele de înaltimea echivalenta a talerului teoretic (H) si se calculeaza prin raportarea lungimii coloanei la numarul de talere teoretice. Cu cât aceasta înaltime este mai mica, eficienta cinetica de separare a coloanei va fi mai mare.
Parametrii N si H (ca si cei efectivi Nef si Hef, care se refera la dispersia picului în faza stationara) se pot utiliza pentru a compara eficienta de separare a diferitelor coloane. Ele sunt deduse pe baza teoriei talerelor în cromatografie, însa nu ne dau nici un fel de informatii despre fenomenele care se produc în coloana si parametrii care infuenteaza aceste fenomene. Pentru a explica aceste fenomene, în special rolul proceselor difuzionale în cromatografie, a fost elaborata o alta teorie, teoria cinetica. Teoria cinetica explica dispersia picurilor cromatografice în timpul deplasarii prin coloana, considerându-le drept rezultatul a trei procese:
- difuzia turbulenta
- difuzia moleculara
- rezistenta la transferul de masa
Corelarea acestor procese este data de ecuatia lui Van Deemter, care a fost dedusa pentru cromatografia de gaze, înainte ca cromatografia de lichide sa fi fost raspândita pe scara larga. Aceasta ecuatie ne da corespondenta dintre înaltimea echivalenta a talerului teoretic si viteza fazei mobile si poate fi scrisa în forma restrânsa astfel:
![]() (63)
(63)
Cei trei termeni ai ecuatiei reprezinta contributia la înaltimea echivalenta a talerului si implicit la largirea picului cromatografic a celor trei factori mentionati: difuzia turbulenta, difuzia moleculara si rezistenta la transferul de masa. Aceasta relatie este valabila în principiu pentru toate sistemele cromatografice. În cromatografia de lichide, rolul principal în dispersia zonei îi revine rezistentei la transferul de masa si termenul de rezistenta la transferul de masa contine doua asemenea rezistente, una in faza mobila si una în faza stationara. Asadar fata de cromatografia de gaze apare în plus termenul de rezistenta la transferul de masa în faza mobila, care are un rol foarte important în dispersia zonei cromatografice. Ecuatia Van Deemter pentru cromatografia de lichide se scrie în forma generala astfel:
![]()
unde CS si CM repezinta coeficientul de rezistenta la transferul de masa în faza stationara, respectiv în faza mobila.
La deplasarea fazei mobile în coloana avem mai multe cauze care determina diferente în ce priveste rezistenta la transferul de masa din faza mobila pe faza stationara si invers. Majoritatea fazei mobile este în miscare, însa exista zone stagnante care se gasesc fie adsorbite pe suprafata granulelor de faza stationara fie în interiorul porilor (Figura 4.3).
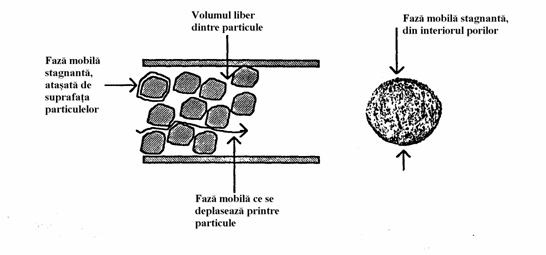
Figura 4.3. Rezistenta la transferul de masa în umpluturi poroase
Solutul ajunge la suprafata fazei stationare trecând prin filmul de faza mobila care stagneaza. Relatia matematica ce reda componenta de rezistenta la transferul de masa pentru acest proces de difuzie în interiorul particulei este complicata, esential fiind însa faptul ca contine termenul dp2.
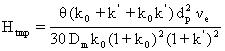 (65)
(65)
sau mai simplu: 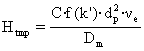 (66)
(66)
unde θ este o constanta care depinde de structura poroasa a particulei, k0 este raportul dintre volumul interior al particulei si volumul liber dintre particule, νe este viteza de curgere a fazei mobile în spatiul dintre particule, iar Dm este coeficientul de difuziune în faza mobila.
Asadar rezistenta la transferul de masa si deci contributia la largirea zonei este proportionala cu patratul diametrului particulei. Pentru a avea eficienta cinetica ridicata în procesele cromatografice de lichide, este neaparat necesara folosirea unor umpluturi cu diametre mici de particule. În acelasi timp, aceasta componenta difuzionala este invers proportionala cu coeficientul de difuziune în faza mobila, ceea ce înseamna ca va fi avantajoasa folosirea unor solventi cu vâscozitate cât mai mica.
Influenta diametrului particulei asupra înaltimii echivalente a talerului teoretic se poate observa si din Figura 4.4, care reda graficul ecuatiei Van Deemter pentru diferite dimensiuni de particule. Cu cât dp este mai mic si în consecinta H mai mic, cu atât eficienta de separarea a coloanei va fi mai ridicata si sansele de a separa compusi având proprietati fizico-chimice asemanatoare vor fi mai mari.
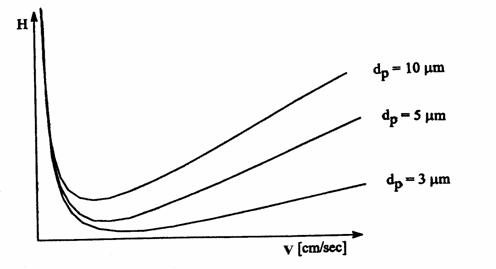
Figura 4.4. Influenta diametrului particulelor asupra ecuatiei Van Deemter
Diferenta de eficienta dintre coloanele cu diametre diferite ale particulelor este importanta atunci când valoarea lui H este determinata în cea mai mare parte de termenii A si C, adica la viteze mari ale fazei mobile. Asadar din punct de vedere analitic ar fi indicata folosirea unei umpluturi cu diametru mic la viteze mari ale fazei mobile, când influenta teremului C·v este determinanta, însa posibilitatea modificarii acestora este limitata din cauza pierderilor de presiune pe coloana cromatografica.
Pierderea de presiune pe coloana este data de urmatoarea relatie:
![]() (67)
(67)
unde φ reprezinta rezistenta coloanei la curgere, iar η este vâscozitatea eluentului. Asadar pierderea de presiune este direct proportionala cu lungimea coloanei, vâscozitatea eluentului si viteza fazei mobile si invers proportionala cu patratul diametrului particulelor de umplutura.
Daca se utilizeaza coloane de lungimi mici, pierderea de presiune poate fi redusa, însa si reducerea lungimii coloanei se poate face doar pâna la o anumita limita (ea influenteaza nefavorabil eficienta coloanei exprimata prin numarul de talere teoretice). O alta problema este ca, lucrându-se cu umpluturi cu diametre mici de particule la debite mari, apare un gradient termic atât pe lungimea cât si pe sectiunea coloanei. Cresterea de temperatura pe lungimea coloanei poate fi foarte importanta ajungând la 20-30ºC, ceea ce modifica vâscozitatea eluentului si poate crea o serie de alte probleme, cum ar fi largirea picurilor sau asimetria acestora. Pe baza acestor considerente, se poate afirma ca în conditiile tehnice actuale diametrul de 2 μm al particulelor de umplutura reprezinta acea limita la care se atinge eficienta cinetica maxima, fara a afecta functionalitatea coloanei.
O alta posibilitate de a mari eficienta de separare a coloanei ar fi cresterea lungimii coloanei, tinând cont de faptul ca numarul de talere teoretice este proportional cu patratul lungimii coloanei. Folosirea unor coloane mai lungi de 30 cm este însa limitata de faptul ca umplerea uniforma a unei coloane lungi este dificila de realizat din punct de vedere practic, iar o umplutura neuniforma va avea o eficienta mai scazuta datorita cresterii termenului difuzional A din ecuatia Van Deemter. Pe de alta parte, s-a vazut ca si pierderea de presiune în coloana creste odata cu cresterea lungimii acesteia. S-a constatat însa ca la legarea mai multor coloane cu umplutura de dimensiuni mici în serie, în anumite cazuri s-a obtinut o însumare a numarului de talere teoretice, dar numai când coloanele au fost foarte asemanatoare, pentru ca proprietatile de curgere prin aceste coloane sa nu fie diferite.
Un alt parametru de eficienta a separarii este rezolutia RS. Relatia de calcul a rezolutiei, care coreleaza toate tipurile de interactiuni care influenteaza separarea, este:
![]() (68)
(68)
În aceasta relatie, atât numarul de talere teoretice cât si factorul de capacitate sunt calculate pentru componenta cu timp de retentie mai mare. Primul termen al acestei relatii ne arata dependenta rezolutiei de factorii cinetici (dispersia picului cromatografic), cel de-al doilea termen de factorii termodinamici (exprimati prin coeficientul de separare) iar cel de-al treilea termen de factorii care determina retentia componentei. Pentru ca o separare sa fie corespunzatoare trebuie ca:
α > 1,05
1< k' < 10 (optim 2 < k' 5)
N > 12000
În aceste conditii, rezolutia minima a doua componente trebuie sa fie de cel putin 1 (preferabil 1,5). În cromatografia de lichide se realizeaza eficiente înalte de separare ale coloanelor, mai ales daca diametrul particulelor de umplutura este mic, ajungându-se curent la un numar de talere de 100.000 sau chiar mai mult. Aceasta înseamna ca se pot atinge si rezolutii ridicate ale separarilor.
Primul parametru care a fost utilizat pentru identificare în cromatografia de lichide a fost volumul de retentie ajustat VR'. Exista însa o problema majora privind utilizarea acestui parametru, aceea legata de determinarea volumului mort. Volumul de retentie ajustat este diferenta dintre volumul de retentie si volumul mort, însa volumul mort (VM) ar trebui sa fie doar volumul fazei mobile din coloana si nu volumul total de lichid care se gaseste între injector si detector. Volumul mort total (VMt) va include si volumele aferente injectorului, tubulaturii de conectare între injector, detector si coloana si celulei de masura. El se poate determinat doar utilizând un solut care nu se adsoarbe pe faza stationara si care are aceeasi marime moleculara cu cea a solutului analizat pentru a avea aceleasi interactiuni de excluziune. Daca însa volumul de retentie ajustat este mult mai mare decât volumul mort total, atunci orice solut care nu se adsoarbe poate fi utilizat pentru determinarea volumului mort si în acest caz volumul de retentie ajustat poate fi utilizat drept parametru calitativ. Aceasta utilizare este însa limitata de dependenta volumului de retentie ajustat de conditiile de analiza, mai ales de lungimea coloanei si debitul fazei mobile.
Factorul de capacitate poate fi de asemenea utilizat pentru scopuri de identificare, însa si el depinde de volumul mort, fiind în realitate exprimat în functie de volumul mort total întrucât acesta este cel determinat experimental. Este mai potrivit pentru analiza calitativa decât volumul de retentie ajustat, însa are dezavantajul ca depinde de raportul fazelor, care difera de la coloana la coloana.
Utilizarea coficientului de separare α are avantajul ca elimina efectul erorilor de identificare datorate rapoartelor de faze diferite si debitelor de faza mobila diferite care pot apare la trecerea de la o coloana la alta. În cromatografia de lichide, coficientul de separare se poate exprima si ca raportul volumelor de retentie ajustate a doi soluti prin relatia:
![]() (69)
(69)
Daca ignoram efectele de excluziune si consideram ca marimile moleculelor celor doi soluti sunt apropiate, rezulta ca coeficientul de separare va fi egal cu raportul coeficientilor de distributie, adica va fi independent de raportul fazelor si de debitul fazei mobile. Pentru a usura obtinerea unor valori mai reproductibile care sa fie utilizate în scopul identificarii calitative, de multe ori în sistem se introduce o substanta standard si coeficientul de separare al tuturor componentelor probei de analizat este exprimat fata de acest standard. Valorile astfel calculate reprezinta de fapt retentiile relative (r) fata de standardul ales.
Trebuie însa precizat ca nici una dintre metodele prezentate bazate pe retentie nu poate duce la identificari calitative certe si lipsite de ambiguitati. Pentru aceasta este nevoie si în cazul cromatografiei de lichide de utilizarea unor metode auxiliare de identificare, dintre care cele mai bune sunt cele spectrometrice: IR, RMN, masa. Exista la ora actuala sisteme LC-MS sau LC-IR care permit o analiza calitativa lipsita de erori. De multe ori este foarte utila si confirmarea omogenitatii picului cromatografic, adica a faptului ca picul respectiv reprezinta semnalul doar a unei singure componente. Acest lucru poate fi realizat prin folosirea unui detector cu sir de diode.
4.3.1. Cromatografia de adsorbtie lichid-solid
Cromatografia lichid-solid (LSC), numita si cromatografie de adsorbtie, utilizeaza o faza mobila lichida si o faza stationara solida formata dintr-o pulbere poroasa având capacitate mare de adsorbtie si o suprafata specifica ce variaza într-un domeniu foarte larg, între 50-1000 m2/g. Acest tip de cromatografie se bazeaza pe interactiunea moleculelor solutului cu centrii activi aflati pe suprafata unui adsorbent solid care reprezinta faza stationara. Adsorbentul se poate gasi fie într-o coloana (în cazul cromatografiei pe coloana) fie pe suprafata unei placi (în cazul cromatografiei în strat subtire). Granulatia umpluturii variaza pe un domeniu larg, de la 5 μm în cazul celor folositi în HPLC si pîna la mai mult de 100 μm în cazul celor folositi în cromatografia pe coloane clasice.
Cromatografia lichid-solid poate fi utilizata cu rezultate bune atât pentru separarea compusilor polari cât si a celor nepolari si este folosita cu precadere în separarile cantitative ale amestecurilor de substante organice, atunci când aceste amestecuri nu sunt foarte complexe. În general, compusii care se pot separa cel mai bine prin tehnica LSC sunt cei care au caracter neionic si sunt solubili în solventi organici. Compusii neionici solubili în apa se separa mai bine prin cromatografie de lichide pe faza inversa sau cromatografie cu faze chimic legate.
Din punct de vedere istoric, a fost prima tehnica cromatografica utilizata (Ţvet).
Teoria echilibrului de adsorbtie în cazul LSC se bazeaza în cea mai mare parte pe izotermele de adsorbtie de tip Langmuir. Aceste izoterme sunt presupuse a fi specifice adsorbtiei moleculelor plane sau orizontale, aria necesara adsorbtiei unei molecule în acest caz fiind mai mare decât la adsorbtia în configuratie verticala. Acest lucru influenteaza cantitatea de proba necesara a fi adsorbita pentru formarea unui strat monomolecular.
Expresia matematica a izotermei Langmuir pentru adsorbtia din faza lichida este de forma:
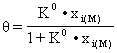 (70)
(70)
unde θ este fractia molara de acoperire a suprafetei adsorbentului de catre componenta adsorbita, K0 este constanta termodinamica a procesului de adsorbtie, iar xi(M) este fractia molara a componentei respective în faza mobila.
În majoritatea cazurilor, suprafata adsorbentului este complet acoperita de un strat monomolecular format din moleculele componentei i si moleculele solventului S. La adsorbtia lichid-solid, exista întotdeauna o competitie între moleculele probei si moleculele solventului pentru ocuparea unui loc de pe suprafata adsorbentului.
Daca se considera stratul monomolecular de solvent drept o faza lichida distincta adiacenta la suprafata adsorbentului, atunci echilibrul de adsorbtie se va putea scrie ca o simpla repartitie a componentei i între cele doua faze lichide:
![]()
unde indicii (M) si (a) reprezinta starea neadsorbita si respectiv adsorbita.
Constanta de echilibru a acestei distributii reprezinta coeficientul de repatitie si este dat de:
![]() (71)
(71)
x reprezentând fractia molara a componentei i în faza adsorbita, respectiv faza mobila.
Constanta termodinamica de distributie înte cele doua faze este data de relatia:
![]() (72)
(72)
ai(a) si ai(M) fiind activitatile respective.
La concentratii mici se poate considera ca Kx = K0. Din punct de vedere practic este însa mai comod sa definim un alt coeficient de repartitie bazat pe concentratii molare, conform relatiei :
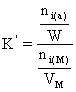 (73)
(73)
unde:
ni(a) reprezinta numarul de moli de compus i adsorbit, raportat la cantitatea de adsorbent W (grame)
ni(M) reprezinta numarul de moli de compus din faza mobila, raportat la volumul fazei mobile VM (ml).
Volumul de retentie VR si migrarea relativa Rf se exprima în cromatografia de adsorbtie cu ajutorul relatiilor:
![]() (74)
(74)
![]() (75)
(75)
Valoarea factorului de capacitate k' va fi în acest caz:
![]() (76)
(76)
4.3.1.2. Faze stationare utilizate în cromatografia lichid-solid
În cromatografia LSC se folosesc drept faze stationare o serie de materiale pulverulente, dintre care cele mai utilizate sunt alumina si silicagelul. În masura mai mica se folosesc silicatul de magneziu, pamântul de diatomee, cât si o serie de alti adsorbenti: amidon, inulina, fosfat de calciu, carbonat de calciu, hidroxid de calciu. Acestea din urma se utilizeaza numai pentru cazuri speciale si deoarece se prezinta sub forma de pulberi fine trebuie amestecate cu pamânt de diatomee pentru a permite trecerea fazei mobile.
Un anumit adsorbent poate manifesta variatii mari ale suprafetei specifice si ale energiei superficiale în functie de metoda de preparare, modul de activare si gradul final de dezactivare. Aceste diferente se vor regasi si în valorile parametrilor de retentie cromatografici. Suprafata acestor adsorbenti este în general eterogena, prezentând activitati diferite. Suprimarea pozitiilor active (mai ales în ce priveste formarea legaturilor de hidrogen, dar si a centrilor acizi sau bazici) sau dezactivarea prin acoperirea selectiva a acestora duce la cresterea capacitatii liniare de adsorbtie. În cele mai multe cazuri pentru dezactivare se foloseste apa, deoarece s-a constatat ca cele mai bune rezultate se obtin când apa acopera în monostrat 50-100% din suprafata solidului, ceea ce înseamna 0,04 g apa la 100 m2 suprafata (sau 4-15% apa fata de cantitatea de adsorbent). Ca dezactivator se mai pot folosi alti compusi puternic polari ca etilenglicolul sau glicerina.
Pentru obtinerea unui adsorbent de o anumita activitate reproductibila trebuie sa fie parcurse urmatoarele etape:
alegerea tipului de adsorbent;
îndepartarea apei prin încalzire timp de 8-16 ore la aer (125-250ºC pentru silicagel, 200-400ºC pentru alumina);
adaugarea unei anumite cantitati de apa dupa racire, cu care se lasa timp de 8-16 ore pentru echilibrare.
Adsorbentii utilizati în cromatografia LSC se pot prezenta sub doua forme:
granule poroase neregulate, numite faze stationare poroase. Ele au suprafete specifice cuprinse între 100-400 m2/g si au o capacitate de încarcare cu proba de 0,5-1 mg/g.
sfere solide compacte acoperite cu un strat de adsorbent poros, care se numesc faze stationare superficial poroase sau peliculare. Ele au suprafete specifice mult mai mici decât adsorbentii porosi, cuprinse între 5-15 m2/g si pot fi încarcate cu proba la un raport de 0,1 mg/g adsorbent, deci cantitatea de proba pe care o pot separa va fi mult mai mica. Eficienta de separare a adsorbentilor superficial porosi este însa mult mai mare.
Alumina se poate obtine în functie de temperatura de preparare si activare în mai multe forme. Daca activarea se face la temperatura mai mica de 700ºC se obtine alumina γ, în amestec cu alte forme. Daca activarea se face la o temperatura mai mare decât 1000ºC, se obtine alumina α inactiva. Sorturile de alumina cromatografica au în general suprafete specifice cuprinse intre 100-200 m2/g. Diametrul particulelor sortimentelor comerciale de alumina este cuprins între 50-200 μm (70-290 mesh). Aceste dimensiuni permit o asezare uniforma a umpluturii în coloana, atingerea unui debit rezonabil de faza mobila sub influenta doar a gravitatii si atingerea rapida a echilibrelor de distributie ale solutilor între adsorbent si faza mobila.
Alumina normala contine si diferite cantitati de apa, fie provenita din forma hidratata a aluminei, fie sub forma de apa adsorbita. În ce priveste structura poroasa, s-a observat ca ea contine un sistem de macropori de forma cilindrica având un diametru de 27 Å, la care se adauga pori neregulati cu diametre mai mari.
Activitatea de adsorbtie a aluminei depinde se sortimentul realizat. Exista trei forme de baza de alumina, bazica (pH 10), neutra (pH 7) si acida (pH 4) si este importanta folosirea tipului corect într-o anumita aplicatie din cauza unui posibil efect catalitic. Astfel, alumina bazica poate cataliza hidroliza unor esteri, iar cea acida dehidrogenarea unor alcooli, în special a celor tertiari. Activitatea tuturor celor trei forme de alumina este clasificata în cinci grade (scala Brockmann), pe baza continutului de apa. Gradul I, cel mai activ, se obtine prin activare la 300-400ºC timp de mai multe ore, iar celelalte grade prin adaugare de diverse proportii de apa, dupa racire: 3-4% (gradul II), 5-7% (gradul III), 9-11% (gradul IV), respectiv 15-19% (gradul V). Faptul ca gradul I este cel mai activ înseamna ca va retine cel mai puternic compusii polari.
Silicagelul reprezinta adsorbentul cel mai mult utilizat. Ca structura chimica, este un produs de policondensare al acidului ortosilicic. Silicagelul cromatografic se obtine la pH 7, adica în forma neutra. si acest adsorbent se poate obtine în diferite grade, în conformitate cu continutul sau de apa. Gradul I se obtine prin încalzire la 250ºC timp de mai multe ore, iar celelalte prin adaugarea unor cantitati diferite de apa dupa racire: 5% (gradul II), 15% (gradul III), 25% (gradul IV) si 38% (gradul V). În cazul sortimentelor cu continut ridicat de apa se formeaza un film consistent de apa la suprafata adsorbentului, deci aceste separari pot fi considerate mai curând pe baza de repartitie decât de adsorbtie.
Pe suprafata silicagelului se gasesc o serie de grupari silanolice Si-OH. O parte dintre acestea se pot transforma în grupari siloxanice prin eliminarea apei.
![]()
Suprafata specifica a silicagelului este cuprinsa între 200-800 m2/g. Se considera ca singurele centre de adsorbtie pe care le contine silicagelul sunt gruparile hidroxil de la suprafata. Între aceste grupari si moleculele componentelor adsorbite se stabilesc legaturi de hidrogen, moleculele adsorbite având rol de donori de electroni. Exista trei tipuri de grupari silanolice superficiale: vicinale dar nelegate prin legaturi de hidrogen, legate si izolate (Figura 4.5). De asemenea, mai pot exista la suprafata silicagelului grupari silanolice hidratate. Daca temperatura de uscare depaseste 400ºC, se produce o sinterizare a particulelor, determinând reducerea activitatii specifice si a capacitatii de adsorbtie.
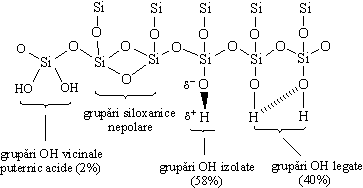
Figura 4.5. Tipuri de grupari silanolice superficiale
Asadar mecanismul adsorbtiei este diferit în cazul silicagelului fata de alumina, unde activitatea de adsorbtie nu se datoreste gruparilor hidroxilice superficiale ci unor interactiuni de natura electrostatica. Acest lucru s-a confirmat prin faptul ca în urma încalzirii la temperatura ridicata, când are loc pierderea gruparilor hidroxilice superficiale, activitatea silicagelului scade iar cea a aluminei creste.
Silicatul de magneziu si Florisilul. Silicatul de magneziu are o activitate de adsorbtie mai mare decât alumina, fiind utilizata pentru separarea unor anumite clase de substante, cum ar fi lipidele. Florisilul este un silicat de magneziu si sodiu cu suprafata specifica de 300 m2/g, iar ca activitate de adsorbtie se situeaza între silicagel si alumina. Prezinta însa si fenomen de chemosorbtie, motiv pentru care utilizarea sa este mai limitata.
Carbunele a reprezentat cel mai popular adsorbent la începuturile cromatografiei, însa la ora aceasta nu prea mai este utilizat.
Dezvoltarea cromatografiei de înalta performanta a determinat realizarea unor adsorbenti cu particule având diametre mai mici de 30μm. Au existat în aceasta directie doua tendinte:
obtinerea unor adsorbenti porosi cu diametrul în jur de 5 μm;
obtinerea unor adsorbenti superficial porosi cu diametrul în jur de 30 μm si având un strat superficial poros cu grosimea de 1-2 μm.
La ora actuala adsorbentii porosi sunt utilizati cu precadere. În cromatografia lichid-solid clasica diametrul particulelor este de pâna la 100 μm, în timp ce în cromatografia în strat subtire clasica, granulatia suportului este cuprinsa între 10-40 μm, utilizându-se ca suport în general silicagel si alumina.
Câteva exemple de adsorbenti pe baza de silicagel poros accesibili comercial, utilizati în mod curent în LSC sunt: Lichrospher (Merck), Porasil (Waters).
În cromatografia LSC se utilizeaza un mare numar de faze mobile lichide, cât si amestecuri ale acestora de compozitie fixa sau compozitie variabila în timp. În cromatografia de lichide, faza mobila poarta numele de eluent, iar procesul de antrenare a solutului prin coloana de catre faza mobila se numeste elutie. Pentru o separare buna, în special în cazul componentelor cu polaritati mult diferite, trebuie utilizata o faza mobila a carei putere de elutie creste. Aceasta putere de elutie este influentata de trei factori:
interactiunile dintre moleculele eluentului si moleculele solutului;
interactiunile dintre moleculele fazei mobile adsorbite si moleculele de solut aflate în faza mobila adsorbita;
interactiunile dintre moleculele de faza mobila adsorbite si adsorbent.
Rolul solventilor în LSC este foarte important, deoarece ei sunt în competitie cu moleculele solutului pentru centrii activi polari ai adsorbentului. Cu cât interactiunile dintre faza mobila si faza stationara sunt mai puternice, cu atât adsorbtia solutului va fi mai slaba si invers. Chiar de la începuturile cromatografiei de adsorbtie s-a constatat ca în cazul adsorbentilor polari solventii nepolari (de exemplu hidrocarburile alifatice saturate sau halogenate) sunt eluenti slabi în comparatie cu solventii polari (alcooli, acizi).
Puritatea solventilor este un aspect foarte important în LSC, deoarece prezenta apei sau a altor compusi polari poate afecta mult performantele coloanei, iar eventuala prezenta a unor compusi care absorb în UV este nedorita atunci când se folosesc detectoare în UV.
Marimea caracteristica pe baza careia se poate stabili faza mobila care trebuie utilizata pentru o anumita separare în conditiile unui adsorbent dat este taria eluentului, care se defineste ca fiind energia de adsorbtie a moleculelor eluentului pe unitatea de suprafata a adsorbentului. Tariile relative, notate cu ε0, au fost determinate pentru o serie de solventi pe diferiti adsorbenti, considerând ca referinta pentanul. În cazul unor amestecuri de eluenti, tariile acestora se pot calcula în functie de fractiile molare si tariile solventilor ce constituie amestecul.
Clasificarea solventilor în functie de taria legaturii lor cu adsorbentul se face în serii eluotrope, care pot fi utilizate pentru gasirea unui eluent cu o tarie potrivita pentru realizarea unei anumite separari. Este însa indicat sa se faca si o testare a eluentului ales si acest lucru se poate face mai rapid prin cromatografie in strat subtire decât prin cromatografie pe coloana. Seria eluotropa reprezinta o serie de solventi cu putere de elutie crescatoare. În cazul adsorbentilor nepolari, taria eluentului se datoreste existentei unor interactiuni nespecifice de tip Van der Waals si creste aproximativ cu masa moleculara a eluentului. Astfel, pentru carbune activ, seria eluotropa în ordinea cresterii puterii de elutie este: apa, metanol, etanol, acetona, propanol, eter etilic, butanol, acetat de etil, hexan. În cazul adsorbentilor polari, taria eluentului creste odata cu polaritatea solventului, deci aproximativ invers decât la adsorbentii nepolari. Astfel, în cazul aluminei, seria eluotropa este data în Tabelul 4.1.
Tabelul 4.1. Seria eluotropa a tariilor relative de solventi pe alumina
|
Solventul |
Taria relativa ε0 |
|
Pentan | |
|
Tetraclorura de carbon | |
|
Toluen | |
|
Benzen | |
|
Cloroform | |
|
Acetona | |
|
Dioxan | |
|
Acetat de etil | |
|
Acetonitril | |
|
Etanol | |
|
Metanol |
Exista serii eluotrope formate din amestecuri de mai multi solventi de polaritati diferite, care pot fi folosite pentru a realiza serii de eluenti de o anumita tarie. Asemenea serii au fost propuse de Snyder si un exemplu, pe silicagel, este dat în Tabelul 4.2.
Tabelul 4.2. Serii eluotrope din amestecuri de solventi, pe silicagel
|
Taria relativa ε0 |
A |
B |
C |
|
100% pentan |
100% pentan |
100% pentan |
|
|
4,2% i-PrCl/pentan |
3% diclormetan/pentan |
4% benzen/pentan |
|
|
10% i-PrCl /pentan |
7% diclormetan/pentan |
11% benzen/pentan |
|
|
21% i-PrCl /pentan |
14% diclormetan/pentan |
26% benzen/pentan |
|
|
4 % eter/ pentan |
26% diclormetan/pentan |
4% acetat de etil/pentan |
|
|
11 % eter/ pentan |
50% diclormetan/pentan |
11% acetat de etil/pentan |
|
|
23 % eter/ pentan |
82% diclormetan/pentan |
23% acetat de etil/pentan |
|
|
56% eter/pentan |
3% acetonitril/benzen |
56% acetat de etil/pentan |
|
|
2 % metanol/eter |
11 % acetonitril/benzen | ||
|
4 % metanol/eter |
31 % acetonitril/benzen | ||
|
8 % metanol/eter |
100% acetonitril | ||
|
20 % metanol/eter | |||
|
50 % metanol/eter |
Se poate observa ca amestecurile de 21% clorura de izopropil/pentan, 14% clorura de metile/pentan si 26% benzen/pentan au aceeasi putere de elutie pe silicagel (egala cu 0,15), de aceea se numesc echieluotropice. Putem avea cazuri de separari când, desi factorii de capacitate au valori potrivite, separarea componentelor, reflectata de valorile coeficientului de separare α, este necorespunzatoare. În asemenea situatii pentru cresterea coeficientului de separare este suficienta de multe ori doar modificarea eluentului prin înlocuirea componentei mai polare, fara a modifica taria eluentului. Acest lucru se poate face folosind un eluent echieluotrop cu primul.
Pot exista si anumite anomalii în comportarea unor solventi, acestea datorându-se unor efecte secundare care nu au legatura cu procesul de adsorbtie, de exemplu formarea unor legaturi de hidrogen între moleculele solutului si eluentului.
În mod uzual, în cromatografia lichid-solid se foloseste un adsorbent polar si elutia se realizeaza la început cu un eluent nepolar, apoi cu tarie crescatoare a fazei mobile, fie prin tehnica elutiei în trepte fie prin cea cu gradient. Nu se recomanda utilizarea unui amestec de solventi în care o componenta foarte polara sa fie prezenta în cantitate prea mica, deorece acesta se va adsorbi preferential si separarea va fi necorespunzatoare. Se poate lucra si cu faze inverse, când se folosesc adsorbenti nepolari cum sunt carbunele activ sau silicagelul cu suprafata silanizata. În aceste situatii, eluentul este constituit dintr-un amestec de apa si un solvent organic miscibil cu apa, iar cresterea tariei eluentului se realizeaza prin cresterea proportiei de solvent organic în acest amestec.
4.3.1.4. Tehnica cromatografiei lichid-solid pe coloana
Cromatografia de adsorbtie este potrivita mai ales pentru separarea compusilor cu mase moleculare medii, polari si nepolari dar fara sarcini electrice.
Cromatografia de lichid-solid se poate realiza prin trei tehnici posibile:
cromatografie lichid-solid clasica, pe coloana;
cromatografie în strat subtire;
cromatografie lichid-solid de înalta performanta.
Dintre acestea, cromatografia lichid-solid pe coloana se utilizeaza practic numai în scopuri preparative, în timp ce cea de înalta performanta se utilizeaza mai ales în scop analitic, dar si preparativ.
Pentru a testa posibilitatea separarii unui amestec de compusi prin cromatografie lichid-solid este indicata cromatografia în strat subtire. În acest scop se foloseste adsorbentul care se preconizeaza a se utiliza în separarea LSC (alumina sau silicagel în majoritatea cazurilor) si prin TLC se poate stabili eluentul sau sistemul de eluenti cel mai potrivit. Pentru realizarea unei separari corespunzatoare sunt importante natura adsorbentului, dimensiunile sale, dimensiunile coloanei si viteza de elutie.
Coloanele utilizate în LSC sunt confectionate în general din sticla, având lungimea cuprinsa între 10-90 cm si diametrul interior între 1-5 cm. Raportul dintre lungimea si diametrul coloanei trebuie sa fie in intervalul 10-20. Aceste coloane pot fi prevazute cu slifuri la ambele capete, pentru a permite atasarea unei pâlnii de separare pe post de rezervor de solvent în partea superioara, respectiv a unui vas de tip Büchner în partea inferioara pentru colectarea fractiunilor eluate (Figura 4.6).
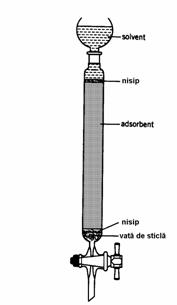
Figura 4.6. Coloana utilizata pentru cromatografia lichid-solid
Tehnica cromatografiei LSC pe coloana consta din trei etape principale:
umplerea si pregatirea coloanei;
încarcarea probei;
elutia componentelor probei.
Umplerea coloanei se poate face prin doua tehnici: uscata si umeda. Tehnica umplerii uscate se utilizeaza mai putin, deoarece necesita cantitati mai mari de adsorbent. Pentru a realiza separari cu o coloana umpluta prin tehnica umeda, este necesara o cantitate de adsorbent de 20-50 de ori mai mare fata de cantitatea de proba care va fi supusa separarii. Daca componentele separate au polaritate apropiata, acest raport trebuie marit la 100-200. Diametrul particulelor de adsorbent utilizate în separarile LSC este cuprins între 0,05-0,5 mm. Particule mai mici duc la o viteza de curgere mult prea redusa prin coloana, în timp ce particule mai mari determina o separare necorespunzatoare. Viteza optima de elutie depinde de multi factori, cum este natura si diametrul particulelor de adsorbent, lungimea coloanei si natura compusilor care trebuie separati. Pentru a realiza umplerea prin tehnica umeda, se realizeaza o suspensie de o parte adsorbent la cinci parti faza mobila si aceasta suspensie este introdusa în coloana. Coloana trebuie sa fie uscata si sa aiba în partea inferioara un opritor din sticla sinterizata, vata de sticla sau nisip, cât si un robinet pentru se a putea regla debitul de elutie. În timpul umplerii coloana trebuie sa se gaseasca în pozitie verticala, iar introducerea suspensiei este dirijata cu ajutorul unei baghete de sticla pentru ca asezarea ei în coloana sa fie uniforma. Dupa introducerea primei portiuni de suspensie, se deschide robinetul pentru evacuarea solventului cu viteza mica si numai dupa aceea se adauga urmatoare portiune. Aceasta adaugare trebuie efectuata înainte ca portiunea precedenta sa se fi asezat complet, altfel umplutura se va aseza în straturi. De asemenea, nu este permis ca nivelul lichidului sa scada sub nivelul stratului de adorbent, acesta trebuind sa se gaseasca permanent în lichid. Dupa umplere, coloana nu are voie sa prezinte goluri, canale, crapaturi sau alte neuniformitati. Dupa terminarea umplerii, peste partea superioara a stratului de adsorbent se pune în general un disc de hâtrie de filtru cu diametrul egal cu diametrul interior al coloanei sau un strat de nisip de 0,5 cm grosime, pentru a preveni dislocarea sa în momentul introducerii probei. Apoi se scurge solventul din coloana pâna când nivelul lichidului ajunge chiar deasupra nivelului umpluturii. În acest moment coloana este pregatita pentru utilizare.
Introducerea probei (Figura 4.7) se face prin aplicarea ei în partea superioara a coloanei, cel mai bine cu ajutorul unei pipete Pasteur. Proba trebuie sa se gaseasca într-un volum minim de solvent, de preferinta acelasi cu cel folosit pentru eluare. Pentru o coloana de 40 cm lungime si 2 cm diametru volumul ideal de solutie de proba este de 1-2 ml. Dupa aplicarea probei se deschide robinetul de la partea inferioara a coloanei si proba este lasata sa intre complet în adsorbent. Apoi cu un volum cât mai mic de solvent se spala peretii coloanei si aceasta portiune de solvent este de asemenea lasata sa intre în adsorbent. Intrarea completa a probei în stratul de adsorbent înainte de începerea elutiei este necesara pentru a preveni diluarea probei. Apoi coloana se umple complet cu eluent si în partea sa superioara se ataseaza rezervorul de eluent. Dupa începerea eluarii, fluxul de solvent prin coloana nu mai este permis sa fie oprit, deoarece ar duce la dispersia excesiva a componentelor.
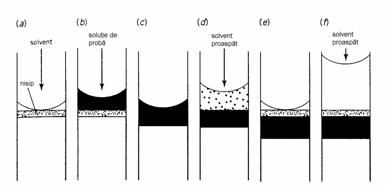
Figura 4.7. Introducerea probei în coloana LSC: (a) nivelul solventului se gaseste chiar deasupra nivelului umpluturii; (b) aplicarea probei; (c) intrarea probei în adsorbent; (d) spalarea peretilor coloanei cu solvent; (e) intrarea acestei solutii în adsorbent; (f) umplerea coloanei cu eluent.
Uneori proba nu este complet solubila în solventul sau amestecul de solventi folosit pentru eluare, dar se dizolva într-un solvent mai polar. Nu este permisa introducerea unei probe incomplet dizolvate, dar nici dizolvate într-un alt solvent mai polar, deoarece ar duce la separari necorespunzatoare. În aceste situatii se recomanda dizolvarea probei în solventul mai polar, adaugarea la aceasta solutie a unei cantitati de adsorbent de 2-10 ori mai mare decât greutatea probei si evaporarea solventului (la un evaporator rotativ) astfel ca proba sa ramâna adsorbita. Acest material se introduce apoi în partea superioara a coloanei si va fi desorbita în timpul trecerii eluentului.
Elutia componentelor se poate realiza în trei moduri:
Elutie izocratica, cu un singur eluent sau cu un amestec de eluenti de compozitie constanta;
Elutie în trepte, utilizând o serie eluotropa de solventi cu putere de elutie crescatoare sau o serie eluotropa de amestecuri de doi solventi, având în general valori ε0 apropiate, pentru separari mai fine.
Elutie cu gradient, utilizând un amestec de doi solventi a carui compozitie este modificata continuu în timpul elutiei.
În cazul în care adsorbentul este polar, alumina sau silicagel, componentele nepolare vor avea o interctiune mai slaba cu adsorbentul si vor elua primele. Elutia izocratica este recomandata mai ales atunci când se urmareste separarea unei componente nepolare dintr-un amestec cu altele mai polare. În acest caz se foloseste un eluent cu polaritate apropiata de cea a componentei urmarite, care va elua doar compusul respectiv, ceilalti compusi, mai polari, ramânând adsorbiti în partea superioara a coloanei.
Daca dupa eluarea componentei sau componentelor mai putin polare vrem sa eluam si componentele mai polare, avem nevoie de un eluent cu tarie mai mare. O asemenea elutie se poate face în trepte, folosind o serie eluotropa de polaritate crescatoare. Nu se recomanda o variatie prea brusca a compozitiei fazei mobile, mai ales daca se face o trecere de la solventi aprotici la solventi protici. Astfel, daca se face trecerea de la pentan la toluen, aceasta se face secvential cu amestecuri de 10%, 50% si 70% toluen în pentan, iar în cazul trecerii de la acetona la metanol numarul treptelor trebuie sa fie mai mare, de exemplu 1, 2, 5, 10, 20, 30, 40, 70 si în final 100% metanol.
Eluarea cu gradient este utilizata atunci când se urmareste schimbarea compozitiei eluentului într-o maniera constanta si uniforma, nu abrupta decât în cazul elutiei în trepte. Acest tip de elutie se poate realiza utilizând doua rezervoare, unul continând solventul mai polar iar celalalt pe cel nepolar (Figura 4.8). Solventul mai polar trece, cu debitul F1, într-o camera de amestecare în care se gaseste solventul mai putin polar. Din aceasta camera, are loc trecerea amestecului de solventi în coloana, cu debitul F2. Se pot genera diferiti gradienti prin variatia celor doua debite. Prin utilizare elutiei cu gradient, se pot separa într-o singura analiza compusi cu polaritati mult diferite.
Regula cea mai importanta în cromatografia de adsorbtie lichid-solid este ca polaritatea (taria) fazei mobile este fie mentinuta constanta în timpul analizei (în cazul elutiei izocratice) fie este crescatoare, dar niciodata ea nu este descrescatoare.
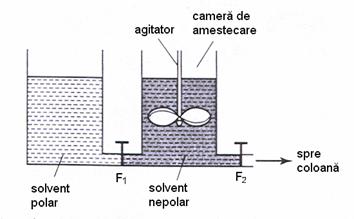
Figura 4.8. Realizarea gradientului de faza mobila
Detectarea componentelor separate în coloana se poate face prin diferite metode. Pentru aceasta, se colecteaza în general fractiuni de volume egale, manual sau utilizând un colector de fractiuni. Aceste fractiuni se analizeaza prin cromatografie în strat subtire, cromatografie de gaze sau spectrofotometrie în UV, apoi fractiunile cu compozitie identica se reunesc si se supun evaporarii pentru indepartarea solventului. Aceasta evaporare se face în general la un evaporator rotativ de vid. Exista si posibilitatea utilizarii unor detectoare UV-VIS automate care se conecteaza la iesirea din coloana si permit monitorizarea componentelor eluate, cu conditia ca acestea sa absoarba în domeniul UV sau vizibil, iar eluentul utilizat sa nu aiba absorbtie (Figura 4.9).
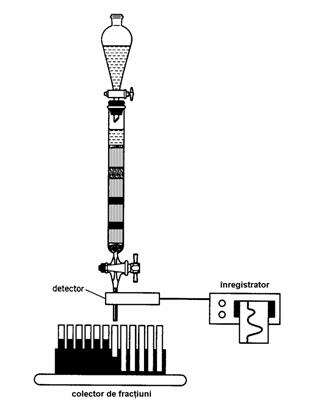
Figura 4.9. Detectarea si colectarea fractiunilor eluate
4.3.2. Cromatografia de repartitie lichid-lichid
Cromatografia lichid-lichid (LLC) are acelasi principiu ca si extractia, adica se bazeaza pe distributia moleculelor solutului între doua faze lichide nemiscibile, în conformitate cu solubilitatea acestora în cele doua faze. Mediul care realizeaza separarea consta dintr-un suport inert (silicagel, kieselguhr) pe care este depus un strat de faza stationara lichida, iar separarea se realizeaza prin trecerea unui flux de faza mobila ce contine proba analizata peste faza stationara. Faza stationara se poate gasi împachetata într-o coloana sau dispusa în strat subtire pe o placa de sticla sau folie de aluminiu.
4.3.2.1. Faze stationare. Faze stationare legate chimic
Cromatografia lichid-lichid se clasifica în functie de polaritatea relativa a fazei stationare si fazei mobile în doua categorii:
Cromatografie lichid-lichid pe faze normale, în care faza stationara este polara iar faza mobila nepolara;
Cromatografie lichid-lichid pe faze inverse, în care faza stationara este nepolara iar faza mobila polara.
În cazul cromatografiei pe faze normale, ordinea de elutie este bazata pe principiul ca componentii nepolari vor avea o preferinta pentru faza mobila si vor elua mai repede, în timp ce componentii mai polari vor avea o afinitate mai mare fata de faza stationara si vor elua mai târziu. În cazul cromatografiei pe faze inverse, ordinea de elutie va fi inversata, în sensul ca primii vor elua componentii mai polari. Utilizarea pe scara foarte larga a cromatografiei pe faze inverse (RPC) la ora actuala se bazeaza pe faptul ca majoritatea compusilor organici au în structura lor regiuni hidrofobe si în consecinta vor putea interactiona cu fazele stationare nepolare. Întrucât faza mobila în cazul RPC contine în mod uzual apa, aceasta metoda este foarte buna pentru si pentru separarea substantelor polare care sunt fie insolubile în solventi organici, fie se leaga prea puternic de adsorbentii polari pentru a putea fi separate prin cromatografie lichid-solid. Un alt avantaj important al cromatografie pe faze inverse este ca solventii utilizati nu trebuie anhidrificati.
Câteva faze stationare tipice si faze mobile utilizate în cromatografia de lichide pe faze normale si faze inverse sunt prezentate în tabelul 4.3.
Tabelul 4.3. Faze mobile si faze stationare în cromatografia lichid-lichid
|
Faze stationare |
Faze mobile |
|
Faze normale |
|
|
β,β'-oxi-dipropionitril |
Hidrocarburi saturate (hexan, heptan), Hidrocarburi aromatice (toluen, xilen), Cloroform, clorura de metilen |
|
Polietilenglicoli (Carbowax) |
|
|
Glicoli (etilen, dietilen) |
|
|
Faze inverse |
|
|
Squalan |
Apa, amestecuri apa-alcool (metanol), Amestecuri apa-acetonitril |
|
Dimetilpolisiloxan |
|
|
Cianoetilsilicon |
|
Desi faza stationara si cea mobila sunt astfel alese încât miscibilitatea lor sa fie minima, chiar si o solubilitate foarte mica a fazei stationare în faza mobila duce la antrenarea ei în timpul curgerii fazei mobile prin coloana. Din acest motiv, faza mobila trebuie sa fie saturata cu faza stationara respectiva înainte de a intra în coloana. Acest lucru se realizeaza în general prin utilizarea unei precoloane (coloane de protectie) situate înaintea coloanei cromatografice. Aceasta trebuie sa contina o umplutura cu granulatie mare (silicagel de 30-60 mesh, de exemplu) pe care sa fie depusa o concentratie ridicata (30-40%) din aceeasi faza stationara care se gaseste si în coloana de analiza.
Suportul fazei stationare este constituit dintr-un material relativ inert, ca de exemplu: alumina, polimeri sintetici (polimetacrilat, copolimer stiren-divinilbenzen), sticla si mai ales silicagel, care are avantajul ca poate fi usor modificat chimic. Este important ca suportul sa nu interactioneze cu solutul, astfel ca mecanismul retentiei sa nu fie unul mixt, de repartitie si adsorbtie ci numai de repartitie. Din acest motiv, atunci când se folosesc adsorbenti porosi drept suport este necesar sa se realizeze o încarcare avansata a suportului cu faza stationara, pentru a acoperi toti centrii activi. Suporturile pot sa fie de doua tipuri: poroase sau superficial poroase. Cele poroase pot fi încarcate cu o cantitate mai mare de faza stationara (între 10-30%), dar eficienta lor de separare este mai scazuta. Suporturile superficial poroase se pot încarca numai cu 0,5-1,5% faza stationara, însa au o eficienta de separare mai mare. Eficienta de separare depinde foarte mult de dimensiunile particulelor de suport, fiind cu atât mai mare cu cât aceste dimensiuni sunt mai mici. La ora actuala în cromatografia de lichide de înalta performanta se utilizeaza suporturi de silicagel total poros cu dimensiuni uniforme, între 3 si 10 μm, de forma neregulata sau sferica. Cele mai eficiente sunt cele sferice (Figura 4.10)

Figura 4.10. Imaginea la microscopul electronic a unui suport cromatografic de silicagel poros
Pentru a elimina problemele legate de pierderea fazei stationare de pe suport în timpul elutiei, se utilizeaza legarea chimica a fazei stationare de materialul suportului. Acest tip de cromatografie de lichide se numeste cromatografie cu faze chimic legate (BPC). Reactiile cele mai mult utilizate pentru obtinerea acestui tip de faze stationare sunt cele de sililare. Ele constau în reactia gruparilor silanolice de la suprafata silicagelului cu clorsilani substituiti. Un exemplu în acest sens este reactia cu un alchil-dimetilclorsilan, în urma careia ia nastere o faza chimic legata monomerica, întrucât fiecare molecula de agent de sililare poate reactiona doar cu o singura grupa silanolica.
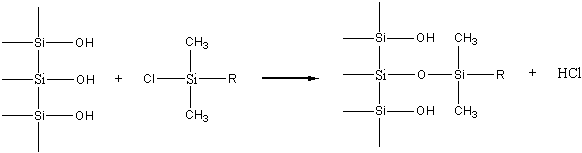
Daca se utilizeaza di- sau triclorsilani în prezenta de umiditate se va forma un strat polimeric pe suprafata silicagelului, adica o faza chimic legata polimerica.
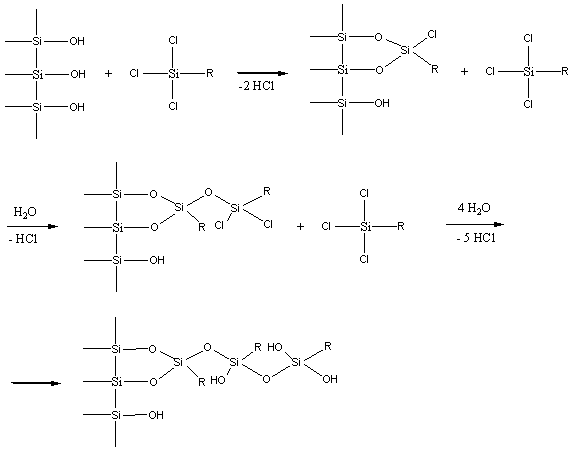
Gruparile silanolice Si-OH vor fi capabile sa adsoarba compusii polari si deci vor afecta proprietatile fazei chimic legate, în general în mod nedorit. Aceste deficiente pot fi eliminate prin inactivarea gruparilor silanolice reziduale prin reactie cu trimetilclorsilan sau hexametildisilazan.
Modul de interactiune al fazei stationare chimic legate cu solutii va depinde de natura gruparii functionale R, cele mai utilizate faze fiind cele nepolare, la care gruparea R este octadecil C-18. Acestea vor fi asadar faze stationare inverse. Alte grupari R de tip nepolar care se folosesc în cazul suporturilor chimic legate sunt cele de tip octil sau ciclohexil. Se utilizeaza si faze stationare chimic legate care au grupari polarizabile, de tip fenil, sau polare, de tip aminopropil, cianopropil sau diol. Cele cu grupari polare vor fi faze stationare normale, utilizate pentru separarea compusilor polari.
O proprietate importanta a fazelor stationare siloxanice este stabilitatea lor ridicata în conditiile de analiza. Ele sunt atacate doar în conditii puternic acide (pH < 2) sau puternic bazice (pH > 9). Un mare numar de asemenea faze stationare destinate analizelor HPLC sunt comercializate sub diverse denumiri.
În cromatografia de lichide, alegerea naturii si compozitiei fazei mobile are o importanta cruciala pentru a obtine o separare eficienta. Puritatea solventilor utilizati este esentiala. Ele nu pot contine alte materiale decât în urme. În cazul utilizarii de detectoare în UV, solventii trebuie sa fie lipsiti chiar si de urme de substante care sa absoarba în UV. De asemenea, prezenta unor particule solide in faza mobila poate determina distrugerea pompei sau injectorului sau înfundarea coloanei. Din aceste motive, solventii cromatografici sunt scumpi si costul lor reprezinta cea mai mare parte a costului unei analize cromatografice. Au aparut sisteme cromatografice care realizeaza recircularea solventului în cazul în care acesta nu contine un solut detectabil, pentru a reduce costurile.
Proprietatea cea mai importanta a solventilor utilizati în cromatografia de lichide este polaritatea. Puterea de elutie a unei faze mobile depinde de polaritatea sa totala, polaritatea fazei stationare si natura componentilor analizati. În cazul separarilor pe faze normale, puterea de elutie creste odata cu cresterea polaritatii solventului, iar în cazul separarilor pe faze inverse ea scade cu cresterea polaritatii solventului. Popularitatea cromatografiei pe faze inverse este determinata în mare parte de avantajele utilizarii unor faze mobile polare ca apa si metanolul: ele sunt mai ieftine, în general mai pure si mai putin toxice decât solventii nepolari.
Desi nu exista o metoda care sa permita determinarea exacta a valorii polaritatii, cea mai raspândita este utilizarea indicelui de polaritate P care reprezinta coeficientul de repartitie al substantei respective între octanol si apa si a fost introdus de Snyder. Acesti indici au fost determinati pe baza unor date gaz-cromatografice si ele sunt accesibile pentru un numar de 81 de solventi. Pe baza indicelui de polaritate solventii se clasifica în 8 categorii. Se considera ca utilizarea indicelui de polaritate este mai adecvata decât varianta mai veche care clasifica solventii în serii eluotrope pe baza tariei relative de elutie a solventului ε0. Indicele de polaritate ia valori între -2 si 10,2 si practic orice valoare în cadrul acestui interval poate fi obtinuta prin amestecarea a doi sau mai multi solventi. Valorile indicelui de polaritate si tariei de elutie pentru câtiva solventi uzuali sunt date în Tabelul 4.4.
Tabelul 4.4. Proprietatile unor solventi utilizati în cromatografia de lichide.
|
Denumirea solventului |
Indicele de polaritate P |
Taria de elutie ε0 (pe oxid de aluminiu) |
|
Fluoroalcani |
< -2 | |
|
Ciclohexan | ||
|
n-Hexan | ||
|
Tetraclorura de carbon | ||
|
Toluen | ||
|
Clorbenzen | ||
|
Eter etilic | ||
|
Clorura de metilen | ||
|
Tetrahidrofuran | ||
|
Cloroform | ||
|
Etanol | ||
|
1,4-Dioxan | ||
|
Metanol | ||
|
2-Propanona | ||
|
Acetonitril | ||
|
Nitrometan | ||
|
Etilenglicol | ||
|
Dimetilsulfoxid | ||
|
Apa |
mare |
Exista si o serie de alte proprietati ale solventilor în afara de polaritate care au importanta în cromatografia de lichide: temperatura de fierbere, vâscozitatea, inflamabilitatea, toxicitatea.
Solventul trebuie întotdeauna filtrat înainte de a fi introdus în rezervorul de solvent al cromatografului. Este foarte importanta de asemenea eliminarea aerului dizolvat sau sub forma de bule din solvent, ceea ce se face prin degazare în vid, prin barbotare de heliu, prin încalzire sau ultrasonare, primele doua metode fiind cele uzuale.
Elutia în cromatografia de lichide de înalta performanta se poate face în doua variante:
elutie izocratica
elutie cu gradient
Elutia cu gradient se utilizeaza mai ales în cromatografia de lichide cu faze chimic legate si în special în cea cu faze inverse. Cele mai mult folosite sisteme binare pentru elutia cu gradient sunt metanol-apa si acetonitril-apa. Dintre acestea, sistemul metanol-apa se foloseste pentru eluarea compusilor mai polari. Daca polaritatea compusilor analizati este foarte asemanatoare, de multe ori este nevoie de folosirea unui sitem ternar de elutie pentru a ajusta mai fin polaritatea fazei mobile. Cel de-al treilea solvent folosit este de multe ori tetrahidrofuranul.
Optimizarea compozitiei fazei mobile
Modificarea compozitiei fazei mobile reprezinta cea mai simpla metoda pentru a influenta rezolutia componentilor în cromatografia de lichide. Pentru separari relativ simple, este posibil ca analiza sa fie efectuata cu un singur amestec de solventi, adica în sistem izocratic. Totusi, gasirea amestecului optim nu este usoara tinând cont de faptul ca în general este vorba despre un amestec de doi (binar) pâna la patru (cuaternar) solventi. Exista o serie de metode pentru determinarea acestui punct optim bazate pe secvente de analize care permit ajustarea compozitiei pâna la atingerea celei optime, pe baza unor algoritmuri de tip simplex. Dezavantajul unei asemenea metode este ca necesita un numar mare de determinari cromatografice. Cea mai buna metoda de determinare a compozitiei optime consta în folosirea unui triunghi (în cazul sistemelor ternare) respectiv a unui tetraedru (în cazul sistemelor cuaternare), compozitia optima fiind data de un punct în interiorul acesti triunghi sau tetraedru (Figura 4.11).
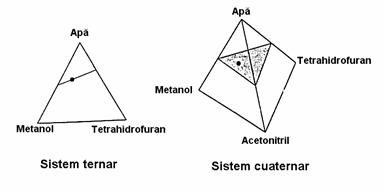
Figura 4.11. Optimizarea compozitiei fazei mobile
Aceste optimizari încep realizarea unor analize cu gradient de elutie de la 0 la 100% pentru perechile de solventi, care duc la stabilirea unei drepte (pentru sistemele ternare) respectiv a unui plan (pentru sistemele cuaternare) în interiorul caruia se determina apoi compozitia optima.
Se pot folosi si metode care sa necesite un numar mai mic de experimente, pe baza unor programe de calculator care sunt accesibile comercial. Asemenea optimizari se practica mai ales în cazul cromatografiei cu faza inversa si ele permit înlocuirea elutiei cu gradient de solvent cu elutia izocratica. Avantajul elutiei izocratice sunt ca este mai rapida, ceea ce este avantajos mai ales în cazul analizelor în serie si ca nu necesita reechilibrarea (revenirea la compozitia initiala) dupa fiecare analiza, ceea ce duce la reducerea timpului de analiza.
4.3.2.3. Aparatura utilizata în cromatografia lichid-lichid
Asa cum s-a aratat, cromatografia de lichide de înalta performanta s-a dezvoltat dupa ce teoria separarilor cromatografice a demonstrat ca pentru obtinerea unei eficiente de separare ridicate, comparabila cu cea realizata în cromatografia de gaze, este nevoie de utilizarea unor umpluturi cu granulatie fina, de ordinul a 5-10 μm. Pentru a avea însa viteze de elutie corespunzatoare, în aceste situatii se impune folosirea unor presiuni ridicate la intrarea eluentului în coloana. Pe de alta parte, folosirea unor coloane scurte a dus la scaderea importanta a capacitatii de încarcare cu proba, ceea ce a necesitat construirea unor detectoare de mare sensibilitate.
Schema de principiu a unui cromatograf de lichide de înalta performanta este data în Figura 4.12:
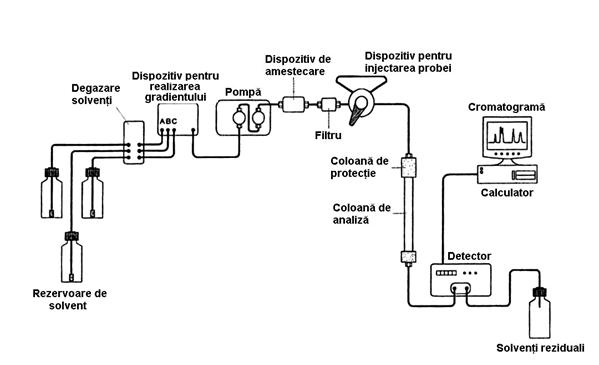
Figura 4.12. Schema unui cromatograf de lichide de înalta performanta (HPLC)
Principalele componente ale cromatografului de lichide sunt:
- sistemul de alimentare cu faza mobila
- dispozitivul pentru introducerea probei
- una sau doua pompe
- coloana
- detectorul
- sistemul de prelucrare a datelor
4.3.2.3.1. Sistemul de alimentare cu faza mobila
Cuprinde urmatoarele dispozitive, care pot fi asamblate în diferite configuratii:
rezervoarele de solventi (Figura 4.13);
dispozitivul pentru degazarea solventilor;
dispozitivul pentru realizarea gradientului de solventi.
Dispozitivul pentru degazare este necesar pentru îndepartarea din solventi a gazelor dizolvate, mai ales a oxigenului, care în timpul trecerii prin coloana ar putea reactiona cu faza stationara, sau ar putea forma bule de gaz cu influenta nefavorabila atât asupra separarii cât mai ales a detectiei, tinând cont de volumul foarte mic al celulelor detectoarelor moderne. Degazarea se realizeaza în doua moduri:
prin barbotare de heliu în rezervorul de solvent;
prin trecerea solventului printr-o unitate de degazare cu vid, unitate prevazuta cu o tubulatura de teflon care permite difuzarea prin pereti a gazelor prezente în solvent, dar nu si a moleculelor solventului (Figura 4.14).
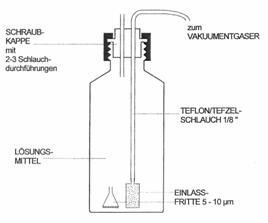
Figura 4.13. Rezervor de solvent utilizat în cromatografia HPLC
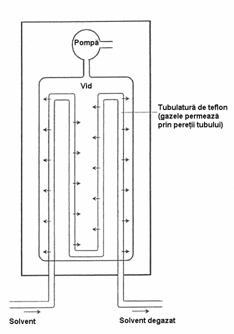
Figura 4.14. Dispozitiv de degazare a solventilor cu vid
Dispozitivul pentru alimentarea cu solventi poate fi foarte diferit ca si complexitate, de la un simplu rezervor de solventi în cazul elutiei izocratice pâna la un programator de eluent pentru doi sau trei solventi.
Prin elutia cu gradient de solvent se poate realiza separarea componentelor care nu se separa prin elutie izocratica. Este foarte eficienta mai ales daca polaritatea componentelor analizate difera foarte mult.
Exista doua tipuri de sisteme de elutie cu gradient de solvent:
Sistemul cu gradient de joasa presiune, în care solventii se preiau din rezervoare diferite într-o camera de amestecare, apoi amestecul de solventi este pompat prin coloana (Figura 4.15). Cromatografele moderne poseda electrovalve de proportionare pentru solventi, comandate de un microprocesor. În acest mod creste rezolutia, se reduce timpul de analiza si creste si sensibilitatea analizei.
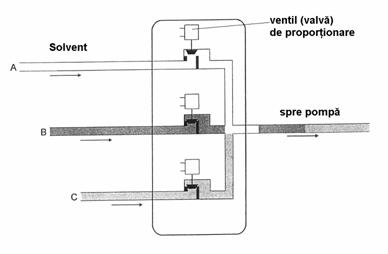
Figura 4.15. Formarea gradientului de solventi cu ajutorul sistemului cu
valva de proportionare
Multe cromatografe de lichide moderne poseda sisteme pentru vehicularea solventilor care pot amesteca doi sau mai multi solventi, progresiv, într-un raport cuprins între 0 si 100% pentru oricare solvent. Se obtine o diagrama de compozitie în functie de timp care poate avea forma liniara, convexa sau concava. Programerea compozitiei eluentului îndeplineste acelasi rol in cromatografia de lichide ca programarea de temperatura în cromatografia de gaze, permitând separarea picurilor neseparate corespunzator, fara a afecta rezolutia celorlalte picuri. Gasirea programului optim necesita timp si elutia cu gradient poate avea si anumite inconveniente. De exemplu, este nevoie de un timp la sfârsitul fiecarei analize pentru a reveni la compozitia initiala a eluentului, iar daca unul din solventi are un semnal mai puternic în detector decât ceilalti, se va înregistra o deriva a liniei de baza (drift). Din acest motiv, pentru separarea unor amestecuri mai simple, se prefera elutia izocratica desi timpul de analiza este mai mare, însa nu se mai consuma timp pentru gasirea programului ideal pentru elutia cu gradient.
Avantajul acestui sistem de gradient este ca se foloseste o singura pompa, desi pâna la urma sistemul de programare cu ventile de proportionare este aproape tot asa de complicat si de scump ca si utilizarea unei a doua pompe.
Sistemul cu gradient de înalta presiune, care opereaza cu câte o pompa pentru fiecare solvent. În general, aceste pompe sunt deservite de motoare pas cu pas, iar debitul pompei este controlat de frecventa de alimentare a motorului respectiv. Acest sistem este mai scump decât celalat, însa reproductibilitatea rezultatelor este mai buna.
4.3.2.3.2. Dispozitive pentru introducerea probei
Introducerea probelor poate fi realizata în doua moduri: prin injectie cu ajutorul unei seringi sau prin folosirea unei valve (robinet) de injectie cu mai multe canale (Figura 4.16). Injectia cu seringi se aplica mult mai putin decât în cromatografia de gaze pentru ca ea necesita seringi rezistente la presiune ridicata si un septum construit dintr-un material care sa nu permita extragerea unor compusi de catre faza mobila, care sa dea nastere unor picuri-fantoma.
Valvele de injectie permit introducerea reproductibila a probelor în coloanele cromatografice aflate sub presiune, fara întreruperea curgerii fazei mobile. Proba este incarcata la presiune atmosferica într-o bucla exterioara a valvei de injectie si este apoi introdusa în faza mobila printr-o simpla rotire a valvei. Volumul de proba introdusa variaza între 2μl si mai mult de 100 μl si poate fi modificat prin schimbarea buclei de injectie sau folosind valve speciale cu volum variabil de proba. Pentru analize în serie se recomanda folosirea unor dispozitive de introducere automata a probelor.
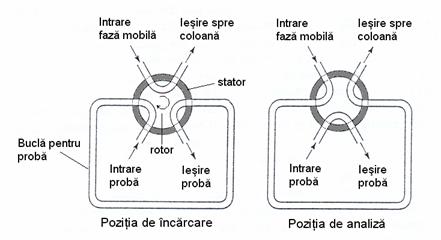
Figura 4.16. Dispozitiv pentru introducerea probei cu bucla de proba cu volum determinat
4.3.2.3.3. Coloana cromatografica
În mod obisnuit, coloanele cromatografice utilizate în analizele HPLC sunt confectionate din otel inoxidabil, au lungimea cuprinsa între 10-30 cm si diametrul interior de 4 sau 5 mm. Pentru retinerea fazei stationare în coloana, la capetele acesteia se gasesc doua frite din otel inoxidabil cu ochiuri de 2 μm sau mai putin.
Umpluturile folosite in cromatografia de lichide moderna au dimensiuni mici, sunt rigide si uniforme. Ele pot fi clasificate în urmatoarele categorii:
Particule polimerice pe baza de copolimer stiren-divinilbenzen. Acestea se folosesc pentru cromatografia de schimb ionic si cromatografia de excluziune sterica, însa au fost înlocuite pentru o serie de aplicatii de umpluturi pe baza de silicagel care sunt mai eficiente si mai stabile mecanic.
Particule superficial poroase (peliculare), cu diametrul cuprins între 30-55 μm, care constau dintr-un învelis subtire (1-3 μm) de silicagel, dispus pe un nucleu dintr-un material inert, de exemplu granule sferice de sticla. Asemenea umpluturi peliculare se mai folosesc în anumite aplicatii de cromatografie de schimb ionic, însa utilizarea lor a înregistrat un puternic regres odata cu dezvoltarea umputurilor total poroase.
Particule total poroase de silicagel, cu diametre mai mici decât 10 μm care sunt cele mai folosite la ora actuala pentru coloanele analitice HPLC. Comparativ cu umpluturile peliculare, ele ofera o serie de avantaje în ce priveste eficienta coloanei, capacitatea de încarcare cu proba si viteza analizei.
Dezvoltarea fazelor stationare chimic legate a avut de asemenea o mare importanta pentru pentru cromatografia lichid-lichid pe coloane de silicagel.
Procedura aleasa pentru umplerea coloanei depinde în mare masura de caracteristicile mecanice ale umpluturii. În cazul umpluturilor cu particule de diametre mai mari de 20 μm se poate folosi tehnica de umplere uscata, în timp ce în cazul celor cu diametre mai mici de 20 μm se face umplere umeda, adica particulele se suspenda într-un solvent si suspensia rezultata se introduce în coloana, sub presiune. În general se prefera achizitionarea unor coloane gata umplute de la firmele de specialitate, având caracteristici definite în cataloagele acestor firme. Lungimea coloanelor este standardizata, fiind de 300, 250, 150, 100 si 75 mm. Coloanele mai lungi vor avea un numar mai mare de talere teoretice, ele fiind considerate standard. Coloanele mai scurte au avantajul unei viteze de separare mai mari si a unui timp de analiza mai redus.
Diametrul standard al umpluturilor folosite în HPLC este de 5 μm. Umpluturile mai fine sunt mai eficiente (reducerea la jumatate a diametrului particulelor determina dublarea eficientei de separare), însa mai scumpe.
Se fabrica o gama larga de coloane analitice, având diametrele interioare cuprinse în intervalul de la 4,6 mm la mai putin de 0,2 mm. Coloanele preparative au diametrul mai mare de 50 mm. În principiu, cu cât diametrul coloanei este mai mare, încarcarea cu proba va fi mai mare, deci se va putea injecta o cantitate mai mare de proba. Pe de alta parte, exista o serie de elemente care fac coloanele mai înguste mai atractive pentru separarile analitice. Astfel, coloanele mai înguste vor necesita un consum mai mic de solvent, determinând economii importante. Un alt avantaj important este ca picul solutului va elua într-un volum mai mic si întrucât raspunsul majoritatii detectoarelor depinde de concentratie rezulta ca înaltimea picului va fi mai mare iar limita de detectie va fi redusa. Aceste avantaje au dus la dezvoltarea unor coloane capilare cu diametrul interior redus pâna la 0,1 mm, însa folosirea acestora creeaza o serie de alte probleme, cum ar fi cele generate de necesitatea de a lucra cu debite foarte mici de faza mobila sau de a reduce foarte mult volumele moarte, de exemplu cele care apar la conexiunea dintre diferitele componente ale aparatului.
În general, pentru coloanele înguste cu diametre între 2 si 0,5 mm se foloseste termenul "microbore", în timp ce coloanele cu diametre mai mici decât 0,5 mm sunt denumite microcoloane. Microcolanele, la rândul lor, se subîmpart în coloane capilare umplute (diametre între 0,04-0,3 mm) si coloane capilare deschise, cu diametre între 3-50 μm. Acestea din urma sunt construite din sticla speciala si din punct de vedere constructiv sunt similare cu coloanele capilare folosite în cromatografia de gaze. Desi folosirea lor necesita o aparatura speciala în ce priveste constructia pompei si a detectorului, care sa opereze la debite de faza mobila foarte mici, de pâna la 1μl/min, ele permit realizarea unor separari cu o eficienta mai mare de 100.000 talere teoretice într-un timp de analiza mai mic de 30 de minute, cu un consum foarte scazut de faza mobila.
Pentru cresterea duratei de utilizare a unei coloane de analiza HPLC se recomanda introducerea unei coloane de protectie (precoloana, guard column). Aceasta este o coloana mica ce se amplaseaza între injector si coloana analitica si previne pierderea eficientei coloanei analitice datorita prezentei anumitor impuritati prezente în proba sau solvent, de exemplu a celor care se adsorb foarte puternic. Un alt rol posibil al acestei coloane este de a satura solventul cu faza stationara si a preveni astfel antrenarea fazei stationare din coloana analitica. Coloanele de protectie sunt umplute cu faza stationara microporoasa sau peliculara. Cele peliculare sunt mai usoare de manipulat la umplere însa trebuie schimbate mai frecvent deoarece capacitatea lor de retinere este mai scazuta.
4.3.2.3.4. Pompe utilizate în cromatografia de lichide
La ora actuala exista un mare numar de pompe care pot fi utilizate în cromatografia de lichide, dintre care unele sunt destinate numai unor aplicatii specifice.
Aceste pompe sunt de doua tipuri:
pompe pneumatice, care sunt pompe cu presiune constanta;
pompe mecanice, care sunt pompe cu debit constant.
Pompele pneumatice sunt actionate sub influenta presiunii aerului. Avantajele lor sunt constructia simpla, costul redus si posibilitatea realizarii unor presiuni foarte mari. Pe ansamblu însa ele sunt inferioare pompelor mecanice, dezavantajul major fiind acela ca viteza de curgere se modifica la schimbarea vâscozitatii eluentului, ceea ce le face neoperabile în cazul sistemelor de elutie cu gradient. Pompele pneumatice se folosesc la ora actuala pentru umplerea coloanelor cu faza stationara prin metoda umeda si, în anumite cazuri, pentru realizarea debitelor mari necesare pentru cromatografia preparativa.
Pompele mecanice sunt utilizate în majoritatea aplicatiilor. Ele sunt pompe cu piston, care elibereaza solventul în mod constant, fara a necesita oprire pentru reîncarcare ca pompele pneumatice. Debitul lor ramâne constant chiar în conditiile modificarii unor caracteristici ale eluentului ca vâscozitatea sau temperatura si de aceea pot fi folosite în combinatie cu detectoarele de mare sensibilitate, întrucât nu duc la modificari ale zgomotului de fond al acestora. Dezavantajul lor principal este ca orice defectiune care afecteaza curgerea eluentului duce la cresterea brusca a presiunii în sistem. Pentru evitarea accidentelor, aceste pompe sunt prevazute cu limitatoare de presiune, care opresc pompa daca se atinge o presiune maxima stabilita.
Cel mai simplu, mai ieftin si drept urmare mai folosit tip de pompa este pompa cu un singur piston (cu miscare alternativa - "reciprocating"). Aceasta a fost prima pompa de presiune înalta folosita pentru coloane cu particule de dimensiuni mici. Schema acestei pompe este prezentata în Figura 4.17.
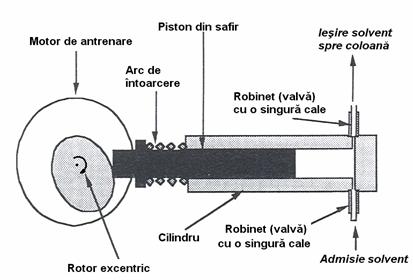
Figura4.17. Pompa de înalta presiune cu un singur piston
Aceasta pompa consta dintr-un piston confectionat din safir, în mod uzual având diametru de 3-4 mm si lungime de aproximativ 4 cm care se gaseste într-un cilindru din otel inoxidabil. Miscarea pistonului este comandata de un rotor excentric care are un profil special ce permite ca miscarea de refulare sa fie liniara si uniforma iar cea de aspiratie neliniara si rapida. Întoarcerea pistonului pentru cursa de aspiratie este determinata de un arc de întoarcere. Intrarea si iesirea solventului din pompa sunt reglate cu ajutorul a doua robinete cu o singura cale, care nu permit deplasarea sa decât într-o singura directie. Volumul de solvent pompat la o cursa a pistonului este în mod uzual în jur de 250 μl, dar poate fi redus pâna la 2-5 μl daca se utilizeaza coloane înguste.
Acest tip de pompa este de uz general si potrivita pentru majoritatea aplicatiilor LC. Din cauza faptului ca în timpul realimentarii cilindrului fluxul de lichid este oprit pentru o scurta perioada, se creeaza niste pulsatii ale debitului de faza mobila. Aceste pulsatii nu influenteaza negativ separarea în majoritatea cazurilor, deoarece coloana contine suficienta faza mobila pentru a atenua pulsatiile. Se poate folosi si un atenuator de pulsatii. Pulsatiile de debit pot determina cresterea zgomotului de fond pentru anumite detectoare, care nu mai pot fi în aceste conditii folosite la sensibilitatea lor maxima. Totusi, în majoritatea cazurilor ele nu reprezinta o problema reala si nu justifica achizitionarea unor pompe lipsite de pulsatii, care sunt mult mai scumpe.
Exista doua variante de asemenea pompe lipsite de pulsatii. Prima este pompa cu doua capete si dublu efect, care a fost realizata prima oara de firma Waters si consta în utilizarea a doua pompe cuplate astfel încât atunci când una este în faza de umplere cealalta sa fie în faza de refulare (Figura 4.18). Acest lucru este asigurat prin proiectarea corespunzatoare a rotorului excentric, care comanda miscarea ambelor pistoane.
Figura 4.18. Reprezentarea schematica a unei pompe cu dublu efect si doua capete
1- piston; 2- rotor excentric; 3- arc; 4- corpul (cilindrul) pompei.
Acest tip constituie una dintre cele mai populare si mai trainice pompe care se fabrica la ora actuala.
Cea de-a doua varianta de pompa fara pulsatii este pompa cu realimentare rapida. Aceasta foloseste de asemenea doi cilindri si un singur piston comun. Diferenta este ca cea de-a doua pompa este folosita numai pentru realimentarea rapida a primei pompe, care realizeaza pomparea fazei mobile in coloana. Aceasta realimentare are loc de aproximativ 100 de ori mai repede decât refularea, motiv pentru care pulsatiile pompei sunt reduse la minim.
Exista si un alt tip de pompa cu piston care se foloseste în cromatografia de lichide, pompa cu membrana. În acest caz, pistonul nu mai este în contact direct cu faza mobila ci doar cu un fluid numit fluid de actionare (driving fluid). Din acest motiv cerintele de etanseitate între piston si corpul pompei nu mai sunt atât de stricte. Schema de functionare a unui asemenea pompe este prezentata în Figura 4.19.
În urma miscarii aspiratoare si respingatoare a pistonului, fluidul de actionare determina o oscilatie alternativa a diafragmei, care se transmite asupra solventului aflat de cealalta parte. În felul acesta, solventul este aspirat din rezervor si trimis în coloana. Suprafata diafragmei este relativ mare, ceea ce face ca miscarea diafragmei sa nu fie ampla si pompa poate fi exploatata la frecventa suficient de ridicata. Pulsatiile cu frecventa ridicata sunt atenuate mai usor de coloana. Cu toate acestea, trebuie mentionat ca pompele cu membrana nu sunt complet lipsite de pulsatii.
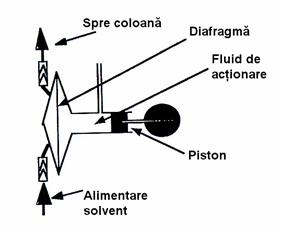
Figura 4.19. Pompa cu diafragma
Utilizarea pompelor trebuie facuta cu multa atentie, nefiind voie ca ele sa functioneze în gol, fara solvent. Utilizarea ca solventi a acizilor clorhidric si bromhidric trebuie evitata deoarece acestia corodeaza chair si otelurile inoxidabile. De asemenea, daca se folosesc ca faze mobile solutii tampon, în special cele care contin cloruri, acestea nu trebuie sa ramâna în pompa dupa terminarea analizei, deoarece pot cauza coroziune.
4.3.2.3.5. Detectoare utilizate în cromatografia de lichide
Functia detectorului este de a monitoriza faza mobila pe masura ce ea iese din coloana. Detectia prezinta mai multe probleme în cromatografia de lichide decât în cea de gaze, neexistând detectoare universale de tipul detectorului cu ionizare în flacara.
Clasificarea detectoarelor se poate face în doua categorii:
Detectoare care masoara modificarea unei proprietati fizice a efluentului coloanei. Asemenea detectoare înregistreaza modificarea unei proprietati fizice a fazei lichide constituite din eluent si componenta separata în coloana comparativ cu eluentul, de exemplu detectoarele de indice de refractie sau de conductivitate. Desi ele au o aplicabilitate destul de larga, au sensibilitate scazuta si sunt afectate de schimbarea compozitiei fazei mobile, deci nu pot fi folosite daca elutia se face cu gradient.
Detectoare care masoara modificarea unei proprietati a solutului. Din aceasta categorie fac parte detectoarele spectrofotometrice, cu fluorescenta si electrochimice. Ele raspund la o proprietate fizica caracteristica numai solutului si în principiu sunt independente de eluent. Au sensibilitate ridicata si un domeniu larg de raspuns liniar, dar datorita specificitatii lor în general este nevoie de mai mult decât un detector pentru a realiza o analiza mai complexa. Exista asemenea unitati care contin mai multe detectoare situate în serie, de exemplu de UV, fluorescenta si conductivitate.
Principalele cerinte pe care trebuie sa le îndeplineasca un detector sunt în buna masura identice cu cerintele pentru detectoarele folosite în cromatografia de gaze:
Sensibilitate ridicata
Domeniu de raspuns liniar cât mai extins
Raspunsul detectorului sa depinda cât mai putin de conditiile de lucru: temperatura si debitul fazei mobile.
La ora actuala exista un mare numar de detectoare folosite în cromatografia de lichide. Cele mai importante, împreuna cu caracteristicile lor, sunt prezentate în tabelul 4.5.
Tabelul 4.5. Principalele detectoare folosite în cromatografia de lichide
|
Denumirea detectorului |
Limita de detectie (μg/ml) |
Elutie cu gradient |
Caracteristici |
|
Spectrofotometric UV-VIS |
DA |
Selectiv, versatil |
|
|
Fluorescenta |
DA |
Selectiv, aplicabil la un numar limitat de compusi |
|
|
Chemiluminiscenta |
2 x 10-7 |
DA |
Selectiv, aplicabil la un numar redus de compusi |
|
Flourescenta indusa prin laser |
Scazuta |
DA |
Selectiv, aplicabil la un numar limitat de compusi |
|
FT-IR |
DA |
Selectiv, versatil |
|
|
Conductivitate |
DA/NU |
Aplicabil pentru ioni si specii ionizabile |
|
|
Amperometric |
NU |
Selectiv, aplicabil pentru compusi cu caracter oxidant sau reducator |
|
|
Indice de refractie |
NU |
Detector universal |
|
|
Spectrometru de masa |
DA |
Detector universal |
|
|
Activitate optica |
NU |
Aplicabil pentru compusi chirali |
|
|
ICP-MS (ICP = inductive coupled plasma) |
2,5 x 10-3 |
DA |
Aplicabil pentru metale |
Exista peste 30 de tipuri de detectoare, cele mai numeroase fiind cele spectrofotometrice, urmate de cele electrochimice si de detectoarele pentru anumite aplicatii specifice. Un aspect important la alegerea detectoarelor este compatibilatatea lor cu colanele înguste sau cele capilare, deorece acestea impun anumite conditii pentru utilizarea detectoarelor.
Detectorul spectrofotometric în UV-VIS
Se bazeaza pe masurarea transmitantei celulei de masura atunci când aceasta este strabatuta de eluentul care contine componentele separate în coloana cromatografica. Conditia necesara este ca aceste componente sa aiba absorbtie suficient de mare în domeniul spectral în care functioneaza detectorul, iar eluentul sa fie transparent în domeniul respectiv. Detectoarele spectrofotometrice în UV-VIS se pot utiliza pentru analiza compusilor aromatici si a celor care poseda legaturi p conjugate: olefine, compusi carbonilici, dervati azoici, nitroderivati, dar si a proteinelor, enzimelor, acizilor nucleici si steroizilor. Ele se pot folosi în elutia cu gradient si sunt aproape insensibile la variatiile de flux date de pulsatiile provocate de pompe. Dezavantajul lor major, alaturi de acela ca substantele analizate trebuie sa absoarba în UV sau vizibil, este ca solventii utilizati trebuie sa fie de puritate deosebita, pentru ca absorbtia lor în UV sa fie nula. Lipsa de absorbtie a solventului este o conditie esentiala mai ales în cazul elutiei cu gradient, deorece nerespectarea acestei conditii ar duce la o deviere continua a liniei de baza. În aceste circumstante, detectorul spectrofotometric poate fi considerat un detector cu raspuns la modificarea proprietatilor solutului. Marimea absorbtiei unei componente este data de cunoscuta lege Lambert-Beer:
![]() (77)
(77)
unde I0 este intensitatea luminii incidente, I este intensitatea luminii transmise, A este absorbanta, ε este coeficientul molar de extinctie (în l/mol·cm), l este lungimea parcursa de raza luminoasa în solutie (în cm), iar c este concentratia componentei în solutie (în mol/l).
Detectorul spectrofotometric în UV-VIS este caracterizat de sensibilitate ridicata, limita detectie fiind 1·10-9 g/ml pentru compusii cu absorbtie puternica si este relativ putin sensibil la variatiile de temperatura si debit ale fazei mobile.
Trebuie mentionat faptul ca aceste detectoare se pot utiliza si în cazul substantelor care nu prezinta absorbtie sau au absorbtie slaba în domeniul de lungimi de unda al detectorului, daca ele se pot transforma în derivati care sa aiba o asemenea absorbtie. Acest lucru se face prin procedeul numit derivatizare postcoloana, în care efluentului coloanei i se adauga un reactiv corespunzator si reactia de derivatizare are loc înainte ca efluentul sa ajunga în detector. Astfel unii aminoacizi, care nu prezinta decât o absorbtie slaba în UV datorata gruparii carbonilice, se pot detecta mult mai usor dupa transformarea în derivati colorati prin reactie cu ninhidrina.
Exista trei tipuri de detectoare în UV-VIS:
Detectorul cu lungime de unda fixa
Detectorul cu lungime de unda variabila
Detectorul cu sir de diode.
Detectorul cu lungime de unda fixa poate fi realizat în varianta cu un singur fascicul sau cu doua fascicule. Detectorul cu doua fascicule este prezentat schematic în Figura 4.20.
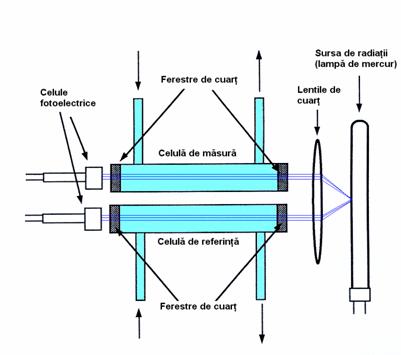
Figura 4.20. Schema detectorului UV-VIS cu dublu fascicul
Lumina provenita de la o sursa de radiatii (în general o lampa de mercur) este focalizata cu ajutorul unui sistem de lentile pe cele doua celule, celula de masura si celule de referinta, apoi ajunge pe doua celule fotolelectrice. Semnalul acestor fotocelule este modificat electronic si apoi reprezinta semnalul de intrare pentru sistemul de achizitii de date al unui calculator sau integrator. Volumul celulei trebuie sa fie cât mai mic cu putinta pentru a reduce dispersia picurilor si a asigura o separare corespunzatoare. Coloanele moderne sunt capabile sa reduca volumul în care elueaza un pic la câtiva μl si daca volumul celulei este prea mare în celula va intra mai mult decât un pic, ceea ce va face imposibila rezolutia picurilor respective. Volumul celulei de masura la un detector modern nu are voie sa fie mai mare decât 5 μl si este recomandat sa fie mai putin de 2 μl. Deoarece absorbanta este proportionala cu lungimea celulei, rezulta ca pentru a avea o celula cu sensibilitate mare ar fi indicata marirea lungimii acesteia. Cresterea lungimii celulei este însa limitata de necesitatea ca volumul celulei sa fie cât mai mic. Pentru a avea o lungime cât mai mare corespunzatoare unui volum cât mai mic, rezulta ca ar trebui redus diametrul celulei, însa si aceasta reducere este limitata pentru ca se reduce concomitent si energia luminoasa care ajunge pe fotocelula si creste zgomotul de fond al detectorului. Celulele detectoarelor moderne au lungimi cuprinse între 1 si 10 mm si diametre interioare de 1 mm sau mai putin.
Detectorul cu lungime de unda variabila utilizeaza drept sursa o lampa care emite o radiatie pe un spectru larg de lungimi de unda (de exemplu o lampa de deuteriu) si prin utilizarea unui monocromator se poate selecta o radiatie cu o anumita lungime de unda dorita, în intervalul 190-350 nm. La unele dintre aceste detectoare, exista si posibilitatea de a opri fluxul de faza mobila, de a retine componenta respectiva în celula si de a-i face spectrul complet în ultraviolet. Uneori aceste spectre pot fi utilizate pentru identificarea calitativa a componentelor respective, desi utilizarea spectrelor în UV în acest scop este limitata. Un alt avantaj al acestui tip de detector este ca pentru fiecare componenta se poate selecta lungimea de unda la care aceasta are absorbanta maxima si astfel de a creste sensibilitatea analizei.
Detectorul cu sir de diode utilizeaza o lampa de deuteriu sau de xenon care emite pe tot domeniul spectrului de UV. Lumina acestei lampi este focalizata cu ajutorul unui sistem de lentile prin celula de masura si apoi ajunge pe o retea de difractie (Figura 4.21). Lumina dispersata de aceasta retea cade pe un sir de diode. Fiecare dioda masoara câte o banda îngusta a spectrului (aproximativ 2 nm), iar masuratoarea are loc într-un timp extrem de scurt (mai putin de 10 milisecunde). Astfel, practic la interval de milisecunde se obtine câte un spectru UV. Utilizarea acestui detector are urmatoarele avantaje importante:
Se obtin spectrele tuturor componentelor separate, care pot fi utilizate pentru identificarea calitativa a acestora.
Prin alegerea unei anumite lungimi de unda se obtine cromatograma în care apar doar compusii care absorb la lungimea de unda respectiva.
Rezultatele unei asemenea analize pot fi prezentate sub forma unor grafice tridimensionale absorbanta-timp-lungime de unda (Figura 4.22). Sensibilitatea detectoarelor cu sir de diode ajunge la 1,5·10-7 g/ml.
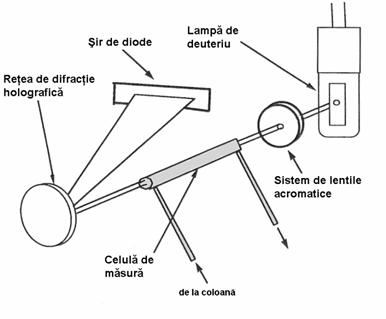
Figura 4.21. Schema detectorului UV-VIS cu sir de diode

Figura 4.22. Cromatograma tridimensionala care prezinta absorbanta în functie de timp si lungimea de unda, realizata cu un detector UV-VIS cu sir de diode
Detectorul cu fluorescenta
Este probabil cel mai sensibil detector folosit in cromatografia de lichide, cantitatea minima detectabila fiind de pâna la 100 de ori mai mica decât în cazul detectoarelor UV-VIS. El poate detecta compusii care prezinta fluorescenta sau pe aceia care pot fi transformati în compusi flourescenti.
Prin fluorescenta se întelege proprietatea unor molecule de a emite o radiatie în momentul în care au atins o stare electronica excitata în urma absorbtiei unei radiatii. Un proces de fluorescenta cuprinde doua etape. În prima etapa, absorbtia unui foton determina trecerea moleculei respective într-o stare electronica excitata. Revenirea într-o stare energetica stabila are loc de asemenea prin emisia unui foton, adica prin ceea ce este numit fluorescenta. Energiile fotonilor absorbiti, respectiv emisi pot varia în functie de nivelele energetice între care are loc tranzitia. În general, emisia se face la lungimi de unda mai mari (adica energii mai mici) decât excitatia. Intensitatea fluoprescentei este proportionala cu concentratia compusului respectiv.
Numarul compusilor care manifesta proprietatea de fluorescenta naturala este destul de redus, în aceasta categorie întrând compusii aromatici, mai ales cei polinucleari condensati. O serie de compusi care nu manifesta fluorescenta naturala pot fi însa transformati în derivati fluorescenti. Aceasta transformare se poate face înaintea separarii cromatografice, numindu-se derivatizare precoloana, sau dupa separarea cromatografica, adica prin derivatizare postcoloana. Derivatizarea precoloana prezinta avantajele ca se pot alege conditiile de reactie pentru derivatizare fara a se tine cont de sistemul de eluent, se poate elimina fara probleme excesul de reactiv de derivatizare, iar în unele cazuri selectivitatea separarii cromatografice pentru compusul derivatizat este mai mare decât pentru cel nederivatizat. De asemenea, nu reprezinta un impediment daca reactia de derivatizare are loc mai lent. Avantajul principal al derivatizarii postcoloana este ca se poate realiza automat, prin dozarea reactivului de derivatizare în efluentul coloanei, fara a fi necesara efectuarea acestei reactii în afara sistemului cromatografic.
Reactivul de derivatizare cel mai mult folosit este clorura de dansil (5-dimetil-amino-naftalin-1-sulfoclorura). Acest reactiv se poate utiliza si în cromatografia în strat subtire, de exemplu pentru analiza aminoacizilor, sau în cromatografia HPLC pentru analiza compusilor farmaceutici. Un alt reactiv de fluorescenta utilizat este dansilhidrazina, care s-a utilizat cu succes pentru analiza aldehidelor si cetonelor.
Un model simplu de detector cu fluorescenta este prezentat în Figura 4.23:
Lampa de UV care emite radiatia de excitatie este în general o lampa de mercur de presiune redusa care emite la 253,4 nm. O serie de substante fluorescente sunt excitate de lumina emisa la aceasta lungime de unda. Radiatia este dirijata cu ajutorul unor lentile prin celula de masura, iar radiatia fluorescenta emisa este focalizata cu ajutorul altor lentile pe o fotocelula.
Exista detectoare de constructie mai complexa, care permit detectarea selectiva a radiatiei fluorescente de o anumita lungime de unda, sau chiar obtinerea unui spectru de fluorescenta al substantei respective.
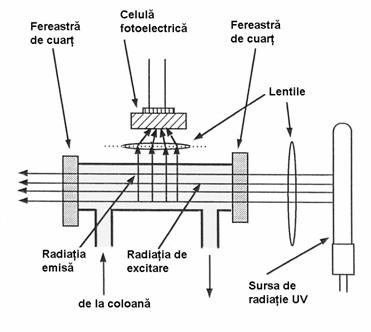
Figura 4.23. Detectorul cu fluorescenta
Detectorul refractometric diferential
Este un detector universal, cu buna stabilitate dar mai putin sensibil, care masoara variatia indicelui de refractie în prezenta componentelor separate în coloana cromatografica. Are si dezavantajul ca este foarte sensibil la schimbarile de temperatura, deci trebuie termostatat cu mare precizie ( 0,001ºC pentru a putea fi utilizat la sensibilitatea maxima). De asemenea, nu poate fi utilizat în cazul în care elutia se face cu gradient si nu se pot folosi decât solventi care au indice de refractie mult diferit de cel al compusilor analizati. Cu toate aceste limitari, detectorul de indice de refractie este extrem de util pentru analiza compusilor neionici, care nu absorb în UV si nu prezinta fluorescenta.
Exista doua tipuri de detectoare refractometrice: cu deflectie si cu reflexie sub unghi limita (de tip Fresnel). Schema de principiu a unui refractometru cu deflectie este prezentata în Figura 4.24.
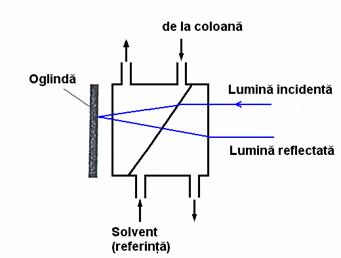
Figura 4.24. Detectorul refractometric diferential
Raza de lumina incidenta, provenita de la un bec cu incandescenta, în general o lampa cu filament de wolfram, trece printr-o lentila de focalizare si este dirijata pe celula detectorului. La trecerea din celula de referinta în cea de masura, raza va fi deviata cu un unghi proportional cu diferenta dintre indicii de refractie ai lichidelor care se gasesc în cele doua celule. Raza reflectata de oglinda trece din nou prin cele doua celule, dupa care este focalizata de o alta lentila pe o celula fotoelectrica. Când raza de lumina îsi schimba pozitia pe celula, semnalul pe care acesta îl emite se va modifica, iar diferenta fata de semnalul de baza este amplificata si constituie de fapt raspunsul detectorului, fiind proportionala cu concentratia componentei în celula refractometrului.
Sensibilitatea acestui detector ajunge pâna la 10-6 g/ml (deci de ordinul ppm). Este utilizat cu rezultate bune pentru analiza hidratilor de carbon si a unor polimeri.
4.3.3. Cromatografia ionica
Cromatografia ionica (IC) reprezinta de fapt versiunea de înalta performanta a cromatografiei de schimb ionic clasice. Poate fi realizata atât utilizând echipamente HPLC uzuale cât si cromatografe construite special pentru aceasta tehnica. Se pot separa atât cationi cât si anioni, folosind rasini schimbatoare de ioni corespunzatoare, însa schimbatorii coventionali au creat probleme pentru separarile HPLC datorita vitezei mici de difuzie prin masa de rasina si datorita compresibilitatii materialului. Aceste probleme au fost rezolvate prin legarea rasinii pe suprafata unor particule sferice de sticla cu diametre cuprinse între 30-50 μm, realizând o umplutura peliculara, sau prin legare de suprafata unor microparticule rigide, realizând o umplutura de tip faza inversa. Asemenea faze stationare pot fi folosite pentru separarea cu rezolutii ridicate atât ale unor amestecuri continând cationi cât si a celor care contin anioni. Eluarea se face cu o faza mobila tamponata si cu tarie ionica crescatoare.
Detectoarele conventionale utilizate în HPLC, cum ar fi cele de UV, pot fi utilizate pentru detectarea ionilor organici, dar nu si a celor anorganici care au necesitat dezvoltarea unor sisteme noi de detectie, printre care s-a impus detectorul de conductivitate electrica. O alta posibilitate este utilizarea detectiei indirecte în UV, când solventul de elutie contine o concentratie constanta dintr-o substanta cu absorbtie mare, de exemple biftalat de potasiu sau benzoat de sodiu, care da un semnal puternic în detectorul de UV. Aparitia în detector a unei alte componente care nu absoarbe în UV va determina scaderea absorbantei si aparitia unui pic negativ în forma originala, care însa prin inversarea polaritatii înregistratorului se poate transforma usor într-un pic obisnuit.
4.3.4. Cromatografia prin excluziune
Cromatografia prin excluziune (SEC) se bazeaza pe separarea moleculelor de solut în conformitate cu marimea lor. Tehnica separarii compusilor solubili în apa si care are loc în solutii apoase este denumita cromatografie prin gel-filtrare si este utilizata de exemplu pentru purificarea proteinelor. Separarile care au loc în solutii neapoase sunt cunoscute sub numele de cromatografie de permeatie prin gel. În ambele cazuri mecanismul separarii este identic si se bazeaza doar pe marimea relativa a porilor fazei stationare comparativ cu marimea moleculelor de solut, neavând loc interactiuni între solut si faza stationara.
Fazele stationare utilizate în acest tip de cromatografie sunt materiale porase cu porozitate strict controlata, iar mecanismul retentiei consta în penetrarea diferita a moleculelor de solut în interiorul particulelor de gel. Moleculele mari nu vor putea patrunde în interiorul gelului si vor fi antrenate de faza mobila prin spatiile dintre particulele retelei de gel. Moleculele mici vor putea patrunde în interiorul gelului, în functie de dimensiunile lor si dimensiunile porilor si deci vor fi retinute mai puternic.
În cromatografia de excluziune sterica clasica, drept materiale pentru faza stationara s-au utilizat dextrani reticulati (Sephadex) sau geluri de poliacrilamida (Bio-Gel). Aceste geluri semirigide nu pot fi însa folosite în conditiile presiunilor ridicate utilizate în tehnicile HPLC, de aceea fazele stationare moderne sunt realizate pe baza de microparticule din copolimer stiren-divinilbenzen (Ultrastyragel), silice, sau sticla poroasa.
Cromatografia prin excluziune are aplicatii analitice atât pentru separarea compusilor organici cât si a celor anorganici. Desi exista asemenea aplicatii si pentru separarea moleculelor simple, ea este utilizata cu precadere pentru separarea biomoleculelor si a altor compusi cu grad ridicat de polimerizare.
Volumul total Vt al unei coloane de excluziune este constituit din trei parti:
Volumul ocupat de particulele solide ale umpluturii, care constituie aproximativ 20% din volumul total.
Volumul porilor Vp, care reprezinta aproximativ 40% din volumul total. Moleculele mici si moleculele de solvent pot trece prin acesti pori în timp ce strabat coloana.
Volumul mort Vm, care reprezinta restul de 40% si este volumul spatiilor dintre particulele de umplutura. Moleculele mari, care nu pot sa intre în pori, vor trece doar prin aceste spatii.
Rezulta de aici ca volumul de elutie util (în care se gasesc componentele separate) este aproximativ jumatate din volumul total al coloanei. O alta observatie importanta este ca o umplutura de anumita porozitate va putea separa efectiv doar moleculele pe un interval de diferenta de masa de 1,5-2,5 ordine de marime. Umpluturile comerciale au marimile porilor cuprinse între 50 Å si 10 Å. Aceste marimi ale porilor nu înseamna dimensiunile efective ale porilor umpluturii din coloana ci dimensiunea minima a standardului de polietilena care nu este retinut, adica limita de excluziune a umpluturii respective.
În cromatografia de excluziune este uzual sa se cupleze mai multe coloane pentru a îmbunatati separarea. Daca se cupleaza doua sau mai multe coloane cu aceeasi limita de excluziune se urmareste cresterea numarului de talere, adica rezolutia, fara a modifica domeniul de masa al compusilor care pot fi separati. Daca se cupleaza doua sau mai multe coloane cu porozitati diferite, se urmareste extinderea domeniului de masa al compusilor care pot fi separati. O serie de coloane comerciale sunt umplute utilizând un anumit solvent si ele trebuie pastrate sub acelasi solvent. Eluentii cei mai utilizati sunt toluenul, tetrahidrofuranul, diclormetanul, cloroformul, dimetilformamida si dimetilsulfoxidul.
Pentru alegerea tipului de cromatografie pentru o anumita separare este necesara cunoasterea caracteristicilor fizice ale amestecului respectiv. La mase moleculare mai mari de 2000, metoda de separare este cromatografia prin gel-filtrare sau permeatia prin gel. În cazul separarii compusilor cu mase moleculare mai mici de 2000, alegerea metodei celei mai potrivite se poate face conform schemei prezentate în Figura 4.25.
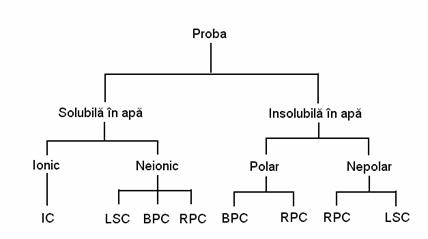
Figura 4.25. Alegerea sistemului cromatografic: IC = cromatografie ionica; LSC = cromatografie lichid-solid; BPC = cromatografie cu faze chimic legate; RPC = cromatografie cu faze inverse.
Trebuie însa mentionat ca o anticipare corecta a metodei cromatografice potrivite nu se poate face cu certitudine si ea trebuie confirmata de experimente, iar în cazul amestecurilor complexe de multe ori este necesara o combinare a mai multor tehnici.
|
