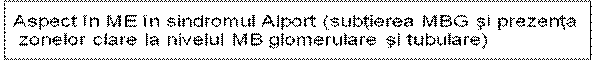Nefropatii ereditare
Risk factors for CKD
|
Non-modifiable |
Modifiable |
|||
|
Family history |
Cadru nosologic n Nefropatiile ereditare ocupa un loc important in practica medicala (10-15% din cauzele BCR). n Numeroase nefropatii cronice primitive sau secundare au la baza, cu o pondere mai mult sau mai putin importanta, factori genetici. Clasificare n Glomerulopatii ereditare n Nefropatii interstitiale ereditare n Nefropatii tubulare ereditare n Boli renale chistice ereditare n Boli metabolice renale ereditare n Afectari renale foarte rare, cu leziuni diverse (glomerulare, interstitiale, displazice, tumori in sindroame plurimalformative) 1.Glomerulopatii ereditare n Sindromul nefrotic congenital n Sindromul Alport n Boala Fabry Sindromul nefrotic congenital n Clasificare: Ø Sindromul nefrotic congenital primar: Sindromul nefrotic finlandez (denumit dupa teritoriul cu cele mai numeroase cazuri) Scleroza mezangiala difuza Glomeruloscleroza focala si segmentara (GSFS) Ø Sindromul nefrotic congenital secundar: Nefropatia cu pseudohermafroditism (Drash) Sindromul ungie-rotula Ø Sindromul nefrotic congenital secundar: Sifilis congenital Toxoplasmoze congenitale Boala congenitala cu citomegalovirus1 Sindromul hemolitic-uremic Disgenezie gonadala Sindromul nefrotic Asociaza trei semne fundamentale: proteinurie peste 3,5g/24ore (>2,5mg/min), hipoproteinemie sub 30g/l, hipercolesterolemie peste 3g/l. O proteinurie persistenta peste 3,5g/24ore sau ,5mg/min(nefrotica) permite afirmarea unui sindrom nefrotic. Cu cat proteinuria este mai mare, cu atat aparitia manifestarilor clinice ale sindromului nefrotic este mai precoce. Hiposerinemia care rezulta explica majoritatea celorlalte manifestari, edemele fiind definitorii in acest sens. Tipuri de sindrom nefrotic (SN) a) SN pur caracterizat prin: . absenta hematuriei macroscopice . absenta HTA . absenta IRC . frecventa mare la copil b) SN impur asociaza la elementele sindromului nefrotic: . hematurie persistenta . HTA . IRC . frecventa egala la copil si la adult Se intalne te la bolnavii cu afec iuni vasculare sistemice: diabet zaharat, amiloidoza, LES, purpura Henoch-Schönlein. Caracteristici genetice n Transmiterea poate fi: Manifestari clinice n Sindromul nefrotic congenital este prezent inca din primele trei luni de viata n Sindromul nefrotic infantil apare in primul an de viata n Ambele forme pot evolua cu proteinurie congenitala sau infantila, cu agresivitate variabila in evolutie n se asociaza frecvent cu afectarea altor organe, evoluand cu pseudohermafroditism (sindromul Drash), microcefalie, anomalii de dezvoltare somatica. Evolutie si indicatii terapeutice n Evolutie variabila: Ø aparitia precoce in primii ani de viata a bolii cronice de rinichi si poate surveni decesul in lipsa tratamentului ( ex: sindromul nefrotic finlandez) Ø deteriorarea mai tardiva a functiei renale-decada a-II-a sau a-III-a de viata-in cazul glomerulosclerozei focale si segmentare Ø Exista doua categorii de pacienti : corticosensibili si corticorezistenti. Ø Sindromul nefrotic corticosensibil are un prognostic bun, cu evolutie lenta spre BCR, Ø Sindromul corticorezistent evolueaza rapid spre BCR stadiul 5 Sindromul Alport Ø Grup heterogen de nefropatii familiale caracterizate prin: hematurie microscopica si leziuni predominant glomerulare, cu afectare a membranei bazale glomerulare Ø Se pot asocia BCR, surditate si alte anomalii (trombocitopenie, hiperprolinemie, hipoparatiroidism, prezenta de anticorpi anti-tiroidieni) Ø Triada clasica clinica este reprezentata de hematurie, surditate si anomalii oculare, aceasta nefiind prezenta la totii pacientii Caracteristici genetice n In 85% din cazuri transmiterea este autozomal dominanta, X-linkata n Boala afecteaza copii de sex masculin n Gena anormala este mostenita de la mama n Femeile purtatoare pot fi asimptomatice sau prezinta doar hematurie izolata fara a dezvolta BCR. n Mai rar transmiterea este autozomal recesiva n Sunt afectati in mod egal copii de ambele sexe n Gena poate fi mostenita de la oricare din parinti n In ambele cazuri defectul genetic intereseaza colagenul de tip IV (constituit din 3 lanturi alfa, care la randul lor sunt de 6 tipuri)
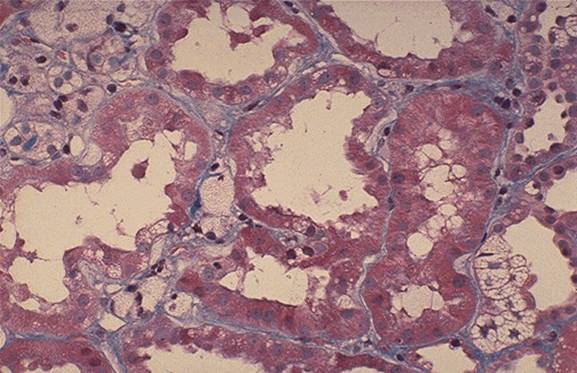
Caracteristici genetice In cazurile cu transmitere autozomal recesiva sunt afectate lanturile alfa-3 si alfa-4, consecinta fiind insa aceeasi : formarea unui colagen IV anormal si a unei MBG fragile. Histologie n MO: initial, hipercelularitate mezangiala, apoi glomeroloscleroza focala asociata cu infiltrate interstitiale n ME: initial, subtierea uniforma a MBG, in stadiile mai avansate apare fragmentarea longitudinala a MBG, ce capata un aspect laminat, cu zone clare in interior care contin granule electonodense, la nivelul MB tubulare proximale si distale n IF: Este negativa sau ofera date nespecifice - depuneri mezangiale granulare de C3 si IgM. Cand se instaleaza scleroza segmentara apar depozite de Ig M, C3, properdina si C4. ³
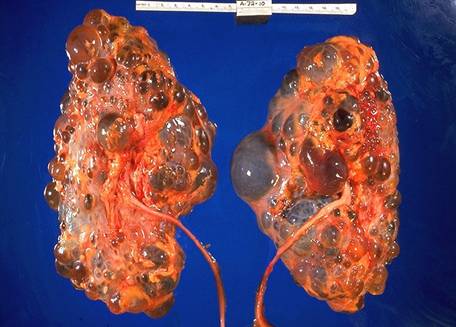
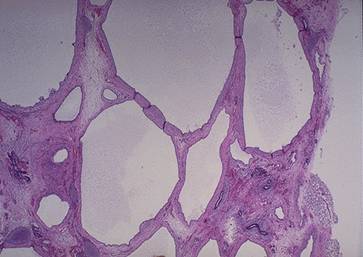
Manifestari Clinice n Renale: Hematurie macroscopica si episoade de hematurie microscopica. Eritrocitele sunt dismorfice si pot aparea frecvent cilindrii leucocitari. Progresiv se instaleaza si proteinuria care poate avea si aspect de sindrom nefrotic (1/3 din cazuri) BCR stadiul 5 se instaleaza lent, obisnuit intre 15-35 ani, mai rar si la 50 ani Clinic n Surditatea Este cea mai frecventa manifestare extrarenala, cu evolutie progresiva, de la o scadere minima a auzului pentru frecventele inalte pana la surditate completa. Leziunile intereseaza predominant stria vasculara si organul lui Corti. n Afectarea oculara Lenticonus anterior: protruzia suprafetei anterioare a lentilelor ochiului, datorata laxitatii colagenului de tip IV, care formeaza capsula lentilei anterioare Diagnostic n Hematuria macroscopica la mai multi membrii ai familiei, asociata cu istoric de de agregare familiala a BCR, la rudele de sex masculin, necesita biopsie cu modificari sugestive atat la pacient cat si la o ruda. n Se suspecteaza sindrom Alport la orice pacient cu hematurie inexplicabila, glomerulopatie sau BCR. n Surditatea este necesara dar nu suficienta pentru diagnostic. Tratament Ø Se aplica masurile de intarziere a progresiei BCR, controlul HTA, restrictie proteica moderata. Ø Transplantul renal se poate complica foarte rar cu aparitia unei GNRP cu Atc anti-MBG. Boala Fabry n Afectiune X-linkata caracterizata prin deficit de alfa-galactozidaza ce determina acumulare de sfingolipide neutre in celulele endoteliale, in podocitele renale si in celulele miocardice. n Barbatii purtatori ai mutatiei exprima afectiunea, femeile pot fi asimtomatice sau prezinta cateva semne ale bolii. Histologic n La nivelul rinichilor glicolipidele se acumuleaza in vasele de sange,in glomeruli si in celulele muschiului neted vascular si in masura mai mica, in epiteliul tubular. n M.O: depunerea lipidica in endoteliul vascular si in celulele epiteliale viscerale glomerulare. Ulterior se produce nefroscleroza. n M.E: Incluzii lipidice grupate, dense in celulele glomerulare, vasculare, tubulare. Clinic n Pot fi interesate: inima, esutul cutanat, sistemul nervos central sau periferic, ochiul, urechea interna, intestinul, rinichiul. n Atingerea renala: proteinurie moderata si evolutie spre BCR stadiul 5, de cele mai multe ori, in timpul celei de-a cincea decada de varsta. n Cea mai precoce manifestare a bolii este defectul de concentrare a urinei. n HTA se instaleaza tardiv. n Manifestari cardiace: cardiomiopatie hipertrofica, tulburari de ritm si conducere, prolaps de valva mitrala, insuficienta mitrala , cardiopatie ischemica. n Manifestari neurologice: parestezii si dureri ale extremitatilor. n Manifestari cutanate: angiokeratoame, hipohidroza. n Manifestari oculare: opacitati corneene, cataracta, anomalii retiniene. Diagnostic pozitiv n Biopsia de piele, PBR examinata la ME confirma diagnosticul. n Determinarea plasmatica a alfa-galactozidazei arata valori mult reduse sau absenta enzimei. Tratament n Tratamentul de substitutie enzimatica arata o reducere a depozitelor tisulare de glicolipide, o incetinire a progresiei BCR sau chiar ameliorare a functiei renale si a hipertrofiei miocardice. n Exista doua preparate de alfa-galactozidaza recombinata umana: Fabrazyme si Replagal, care se administreaza in doze de 1mg/kg, si respectiv 0,2 mg/kg, o data la doua saptamani. 2. Nefropatia interstitiala cronica autozomal dominanta cu hiperuricemie precoce Ø Este o afectiune autozomal dominanta anuntata prin hiperuricemie in timpul copilariei. Ø Clinic: guta juvenila (varsta primei crize de guta este variabila: intre 8 si 32 ani). Ø Uneori hiperuricemia ramane asimptomatica pana in stadiul uremic. Ø HTA este absenta la debut, dar apare in cursul progresiei BCR. 3. Bolile renale chistice n Chisturile apar prin dilatatia unei portiuni din nefron cu formarea unei cavitati acoperite cu epiteliu, pline cu lichid sau material semisolid. n In formarea chisturilor pot interveni: leziuni ale matricei extracelulare inconjuratoare, secretie continua de lichid, hiperplazia epiteliului din peretele chistic. Clasificare 1. Boli ereditare n Afectare cortico-medulara Ø Transmitere autozomal dominanta: - Boala polichistica renala autozomal dominanta - Scleroza tuberoasa - Boala von Hippel-Lindau - Boala glomerulochistica Ø Transmitere autozomal recesiva: -Boala polichistica renala autozomal recesiva n Afectare medulara 2. Boli non-ereditare: - Boala chistica hipokaliemica - Chisturi simple - Boala chistica renala dobandita - Rinichiul spongios medular - Rinichiul multichistic congenital Boala polichistica renala autozomal dominanta (ADPKD) n Este o afectiune sistemica caracterizata prin prezenta de chisturi distribuite difuz in ambii rinichi. n Gena responsabila este localizata pe cromozomul 16 (tipul 1) sau pe cromozomul 4 (tipul 2). n Clinic, cele 2 forme sunt diferite: ADPKD tipul 1 debuteaza mai precoce, iar HTA si IRC apar mai devreme decat in tipul 2. n Boala afecteaza ambele sexe in aceeasi masura, manifestandu-se in decadele 3-4. Anatomie patologica n Macroscopic: rinichii pot atinge dimensiuni foarte mari (pana la 40 cm lungime si 8 kg greutate), avand multiple chisturi pline cu lichid limpede sau sangvinolent. n M.O. evidentiaza chisturile ca diverticuli in capsula Bowman, tubul proximal, ansa Henle si tubul distal. n M.E. a demonstrat ca, pe masura ce cresc, chisturile nu comunica cu tubii de origine.
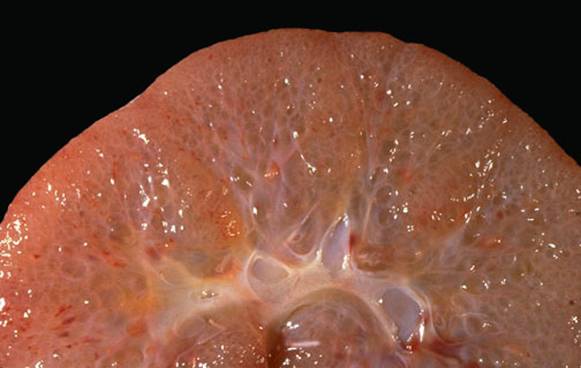
Aspect macroscopic si microscopic (Hematoxilina-eozina) in boala polichistica hepato-renala autozomal dominanta
Clinic Manifestari renale n Durerea abdominala este simptomul cel mai frecvent . Poate fi determinata de intinderea capsulei si tractiunea pediculului renal ( cronic) sau de ruptura unui chist, litiaza sau neoplasm (acut). n Poliuria apare in fazele avansate ale bolii. HTA apare, insa, precoce la mare parte din pacienti, fiind determinata de marirea chisturilor si activarea sistemului renina-angiotensina-aldosteron. n Hematuria poate fi o modalitate de debut frecvent intalnita si cauzata fie de ruptura chisturilor, fie de nefrolitiaza si infectie. n Litiaza renala - pot aparea infectii ale tractului urinar dar si pielonefrite. Diagnosticul diferential dintre cele doua este important deoarece tratamentul este diferit. Pielonefritele evolueaza cu piurie, cilindrii leucocitari, uroculturi pozitive. Tratamentul se face cu antibiotice lipofile. Lipsa de raspuns la terapie dupa 3-4 zile sugereaza prezenta de piochist. Manifestari extrarenale n Chisturile hepatice nu afecteaza functia hepatica, dar compresiunea la nivelul parenchimului hepatic poate necesita interventie chirurgicala. n Chisturile pancreatice sunt intalnite la aproximativ 10% din pacienti si 5% din pacienti au chisturi splenice. n Diverticulii colonici apar cel mai frecvent la pacientii hemodializati. n Afectare cardiaca: prolaps de valva mitrala, insuficienta aortica, HVS secundara HTA. n Anevrismele intracraniene (1-40%) au risc de ruptura mai mare decat in populatia generala. Evolutie. Prognostic n Caracteristic: discrepanta dintre gradul anemiei (usoara) si nivelul crescut al creatininemiei. n Peste jumatate din pacienti evolueaza spre BCR n In cazuri cu infectii severe, recurente, hematurie severa, recurenta sau in cazul suspiciunii unui carcinom renal poate fi necesara nefrectomia. n Chisturile hepatice evolueaza de cele mai multe ori benign n Pacientii cu ADPKD tolereaza bine HD,dar exista un risc crescut al complicatiilor trombotice, ca si necesitatea unei sedinte de hemodializa mai lungi. n Dializa peritoneala este formal contraindicata la pacientii cu dimensiuni foarte mari ale chisturilor Tratament n Durerea: analgezicele pot fi utilizate. Pentru chisturile gigante se poate realiza drenajul percutan, scleroza cu etanol sau tratament chirurgical (aspirarea fluidului si indepartarea chirurgicala a peretului chistic). n Anevrismele celebrale: riscul ruperii lor este maxim la peste 1 cm. Peste aceasta dimensiune se poate lua in discutie sanctiunea chirurgicala. n Hematuria: necesita spitalizare; in absenta infectiei este suficient repausul la pat minimum 48 ore, tratamentul HTA si asigurarea unei diureze peste 2000 ml/zi. Boala polichistica renala autozomal recesiva (RDPKD) n Afectiunea se caracterizeaza prin prezenta de chisturi renale bilaterale fara displazie, ficatul fiind afectat prin fibroza. n Boala se transmite autozomal recesiv si gene responsabila este localizata pe cromozomul 6. n Este evidenta la nastere si frecvent letala in primul an de viata. Anatomie patologica n La nastere rinichi sunt mariti de volum, dimensiunile sunt maxime intre 1 si 2 ani, incepand sa scada pana la 4-5 ani. n Chisturile se dezvolta din portiunea terminala a tubilor colectori n Ectazia ductelor medulare este patognomonica
Clinic n Nou-nascutii pot avea facies Potter cu fante oculare adanci, nas aplatizat, micrognatie, urechi mari. n Majoritatea decedeaza prin sindrom de detresa respiratorie, determinata de hipoplazie pulmonara, sau prin pneumotorax. n Cei diagnosticati dupa primul an de viata prezinta greata, varsaturi, dureri abdominale cu hepatosplenomegalie Diagnostic pozitiv n Se stabileste pe baza: - istoricului familial, - examenului ecografic al tuturor membrilor familiei, - biopsiei hepatice n Ecografia efectuata intrauterin sau neonatal evidentiaza mase renale marite cu margini imprecis delimitate Tratament n Tratamentul este simptomatic. In cazul instituirii HD se recomanda un aport proteic de 1,5-2 g/kg/zi. n HD nu este contraindicata, dar avand in vedere existenta fibrozei hepatice progresive este recomandat dublu transplant hepatic si renal. 4. Afectarea renala in facomatoze n Boala Von Hippel-Lindau n Tumora Wilms (Nefroflastomul) Boala Von Hippel-Lindau n Afectiune autozomal dominanta caracterizata prin prezenta de tumori ce afecteaza cu predilectie sistemul nervos central si retina (hemangioblastom) dar si rinichiul (chisturi sau neoplasme), suprarenala (feocromocitom) sau pancreasul (chisturi sau tumori). n Poate afecta, cu o frecventa variabila si alte organe: pancreas (chisturi, tumori ale celulelor insulare), testicul (tumora a celulelor germinative), ficat, splina, plaman etc. Morfologie n Exista forme de trecere progresiva de la chisturi simple unistratificate pana la neoplasme cu celule clare ale rinichiului. n Chisturile simple se dezvolta mai ales din tubii contorti distali. n Neoplasmele si chisturile atipice provin din tubii contorti proximali. Diagnostic pozitiv - Examenul clinic obiectiv amanuntit - Oftalmoscopia - RMN cerebrala si a coloanei vertebrale - TC abdominala cu substanta de contrast - Ecografie testiculara - Dozarea catecolaminelor urinare - Analiza genetica moleculara Tratament n Tratamentul este chirurgical, variind de la conservator la radical, in functie de numarul leziunilor, caracterul lor uni- sau bilateral, precum si de gradul tumorii. Tumora Wilms (Nefroflastomul) n Neoplasm trifazic embrionar, ce reprezinta cea mai frecventa tumora maligna a aparatului genito-urinar la copil. Clinic Cele mai frecvente semne ale tumorii sunt: La acestea se mai pot adauga hematurie, HTA, ascita, semne determinate de prezenta metastazelor. Paraclinic n Hemograma releva prezenta anemiei. n Biochimic pot exista cresteri ale ureei si creatininei. n Dozarea acidului vanilmandelic, pentru a exclude neuroblastomul. n Ecografia abdominala descrie intinderea tumorii si aspectul rinichiului contralateral. n UIV arata functionalitatea rinichiului. n Radiografia pulmonara si TC ajuta in evaluarea metastazelor. Tratament n In tratamentul chirurgical trebuie respectate: - chimioterapia preoperatorie - incizia larga, pentru a examina intreaga cavitate abdominala - examenul rinichiului contralateral, intraoperator - disecarea cu atentie a vaselor si ureterului si eliberarea lor - excizia tumorii fara a produce ruptura si contaminarea cavitatii abdominale. n Contraindicatii chirurgicale majore: - tumora bilaterala - tumora mare fixata - invazie hepatica Document InfoAccesari: 5720 Apreciat: Comenteaza documentul:Nu esti inregistratTrebuie sa fii utilizator inregistrat pentru a putea comenta Creaza cont nou A fost util?Daca documentul a fost util si crezi ca meritasa adaugi un link catre el la tine in site in pagina web a site-ului tau.
Copyright © Contact (SCRIGROUP Int. 2025 ) |